 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yukari Tsubata | + 1973 word(s) | 1973 | 2021-11-26 09:17:05 | | | |
| 2 | Amina Yu | -75 word(s) | 1898 | 2021-12-08 03:42:52 | | |
Video Upload Options
EGFR mutations are found in approximately 15% of lung adenocarcinomas cases in Europe and the United States, and 55% cases in East Asia. The exon 19 deletion mutation and L858R mutation account for more than 90% of EGFR mutations, making tumors highly sensitive to EGFR-TKIs. Other mutations are rather uncommon, and a few among them, such as G719X, L861Q, and S768I, turn tumors highly sensitive to EGFR-TKIs; in NSCLC patients harboring these mutations, EGFR-TKIs are considered as the main therapeutic agents. On the contrary, patients harboring exon 20 insertion mutations or T790M mutations as primary mutations are resistant to EGFR-TKI and are not indicated for treatment.
1. Introduction
The role of activating mutations in the epidermal growth factor receptor ( EGFR ) gene in non-small cell lung cancer (NSCLC) was reported in 2004 [1]. The development of EGFR tyrosine kinase inhibitor (TKI), as the therapeutic agent, radically overturned the conventional chemotherapy regimen for lung cancer, which was treated with cytotoxic anticancer agents until then [2][3]. Thus, the conventional method of synthesizing numerous candidate compounds and selecting an effective anticancer drug from among them has been replaced by a method of finding a target molecule first and then designing and developing anticancer drugs that specifically inhibit the target molecule. Since then, several molecular targeted drugs have been developed and clinically applied in patients harboring ALK fusion genes, ROS1 fusion genes, BRAF mutations, NTRK fusion genes, and MET mutations [4][5][6][7][8]. Thus, personalized treatment based on the type of genetic mutations harbored by the patients is the new mainstay of chemotherapy for NSCLC.
Genes whose mutations and alterations are directly responsible for the increased growth and progression of cancer are collectively referred to as driver genes. Kinase inhibitors targeting these mutations/alternations are the primary therapeutic choices for patients harboring driver gene mutations. However, the development of drug resistance is one of the most common challenges associated with the use of kinase inhibitors. Patients treated with kinase inhibitors following fulfilling eligibility criteria show remarkable tumor shrinkage; however, despite the high efficacy of these kinase inhibitors, radical cure of stage IV NSCLC remains an unachieved goal because invariably, almost all patients develop acquired resistance within one to several years. Elucidating drug resistance mechanisms and developing new drugs to overcome resistance is indispensable for achieving higher efficacy and radical cancer cure. Osimertinib was the first drug developed to overcome resistance to kinase inhibitors. A third-generation irreversible EGFR-TKI, osimertinib overcomes the drug resistance acquired due to EGFR T790M mutation [9]. Since then, constant efforts have been made to develop drugs for overcoming resistance to kinase inhibitors and cytotoxic anticancer agents. New molecular targeted drugs and their combination with cytotoxic anticancer agents are expected to improve the prognosis of NSCLC patients further. This entry focuses specifically on the latest treatments for EGFR mutation-positive lung cancer and the strategies aimed at overcoming drug resistance.
2. Advances in the Treatment of EGFR Mutation-Positive Lung Cancer
The FLAURA study, a phase-III trial comparing single-agent osimertinib with first-generation EGFR-TKIs (gefitinib or erlotinib) in patients with EGFR mutations (exon 19 deletion mutation and exon 21 L858R mutation) [10], reported that PFS (the primary endpoint) significantly prolonged with osimertinib treatment, 18.9 months vs. 10.2 months (HR 0.46, 95% C.I. 0.37–0.57). Overall survival (OS) also significantly improved with osimertinib, 38.6 months vs. 31.8 months (HR 0.799, 95% C.I. 0.641–0.997). In terms of toxicity such as skin rash and liver damage, osimertinib tends to be milder than first-generation EGFR-TKIs, and osimertinib monotherapy has been established as one of the standard therapies for the first-line treatment of EGFR mutation-positive NSCLC ( Table1 ).
| Trial (Reference) |
Target EGFR Mutation |
Experimental Therapy |
Control Therapy |
PFS 1 (Primary Outcome) | Overall Survival | ||||
|---|---|---|---|---|---|---|---|---|---|
| Months | HR 4 (95% C.I. 5) |
Months | HR (95% C.I.) |
||||||
| Exp 2 | Ctrl 3 | Exp | Ctrl | ||||||
| NEJ002(2) | Sensitizing mutation |
Gefitinib | Cb 6/PTX | 10.8 | 5.4 | 0.33 | 30.5 | 23.6 | - |
| (0.22–0.41) | |||||||||
| WJTOG 3405(3) | Ex19_del Ex21_L858R |
Gefitinib | CDDP 7/DTX 8 | 9.2 | 6.3 | 0.489 | 30.9 | NR 9 | 1.638 |
| (0.336–0.710) | (0.749–3.582) | ||||||||
| EURTAC(13) | Ex19_del Ex21_L858R |
Erlotinib | CDDP | 9.7 | 5.2 | - | - | - | |
| EURTAC(13) | Ex19_del Ex21_L858R |
Erlotinib | Cb/DTX | 9.7 | 5.2 | 0.37 | - | - | - |
| GEM 10 | (0.25–0.54) | ||||||||
| OPTIMAL (14) | Ex19_del Ex21_L858R |
Erlotinib | Cb/GEM | 13.1 | 4.6 | 0.16 | - | - | - |
| OPTIMAL (14) | Ex19_del Ex21_L858R |
Erlotinib | Cb/GEM | 13.1 | 4.6 | (0.10–0.26) | - | - | - |
| LUX-Lung 7 (15) | Ex19_del Ex21_L858R |
Afatinib | Gefitinib | 11 | 10.9 | 0.74 | 27.9 | 24.5 | 0.86 |
| LUX-Lung 7 (15) | Ex19_del Ex21_L858R |
Afatinib | Gefitinib | 11 | 10.9 | (0.57–0.95) | 27.9 | 24.5 | (0.66–1.12) |
| ARCHER 1050 (16) | Ex19_del Ex21_L858R |
Dacomitinib | Gefitinib | 14.7 | 9.2 | 0.59 | 34.1 | 26.8 | 0.76 |
| ARCHER 1050 (16) | Ex19_del Ex21_L858R |
Dacomitinib | Gefitinib | 14.7 | 9.2 | (0.47–0.74) | 34.1 | 26.8 | (0.582–0.993) |
| FLAURA (17) | Ex19_del Ex21_L858R |
Osimertinib | Gefitinib | 18.9 | 10.2 | 0.46 | 38.6 | 31.8 | 0.799 |
| FLAURA (17) | Ex19_del Ex21_L858R |
Osimertinib | Erlotinib | 18.9 | 10.2 | (0.37–0.57) | 38.6 | 31.8 | (0.641–0.997) |
1 PFS, progression free survival; 2 Exp, Experimental therapy; 3 Ctrl, Control therapy; 4 HR, hazard ratio; 5 C.I., confidence interval; 6 Cb, carboplatin; 7 CDDP, cisplatin; 8 DTX, docetaxel; 9 NR, not reached; 10 GEM, gemcitabine.
Meanwhile, several clinical trials have begun exploring the usefulness of osimertinib combination therapy. Combination therapy of osimertinib and CBDCA/PEM in FLAURA2, an international safety run study, exhibited a manageable safety and tolerability profile in first-line settings [11]. In addition, in phase II clinical trial (WJOG9717L) examining the efficacy of bevacizumab (an angiogenesis inhibitor) and osimertinib, the primary endpoint, PFS, did not show a statistically significant prolongation of survival [12]. Further assessment is expected.
3. Mechanisms Underlying Acquired Resistance to EGFR-TKIs
Broadly three mechanisms of acquired resistance to EGFR-TKIs have been reported [13] : (1) alteration of the target via EGFR-TKI susceptibility mutations (acquiring T790M mutation), (2) activation of alternative pathways due to genetic alterations such as MET amplification and KRAS mutation, and (3) alteration of the phenotype, such as transformation to small cell lung cancer or EMT (epithelial-mesenchymal transition). Furthermore, these pathways may be co-activated in a single case, and their crosstalk can further complicate the cancer course and patient outcomes.
Since first-generation EGFR-TKIs were introduced in clinical practice, the molecular mechanisms involved in EGFR-TKI acquired resistance have been vigorously investigated [13][14]. The most frequent mechanism associated with acquired resistance involves the T790M secondary mutation. Under physiological conditions, EGFR , a receptor-type tyrosine kinase, forms a dimer when bound by its ligands such as EGF and TGF-α and transmits pro-proliferation signals in the cells via autophosphorylation and assuming an activated conformation. In EGFR mutation-positive lung cancers, exon 19 deletion mutation and exon 21 L858R point mutation, among others, cause the kinase to become homeostatically activated in a ligand-independent manner [15]. This leads to the transmission of excessive pro-survival and pro-proliferation signals, which in turn lead to cancer initiation and progression; the efficacy of EGFR-TKIs lies in lowering these excessive pro-survival and pro-proliferation signals. When the T790M mutation occurs in the ATP-binding pocket of the kinase, the affinity of the kinase for EGFR-TKI decreases compared to that for ATP, leading to the development of drug resistance [16]. Generally, tumor heterogeneity in which multiple clones with different genotypes are present in one tumor mass has been observed [17]. It is known that in EGFR mutation-positive lung cancer, T790M-positive cells are present at an incidence rate of 79.9%, albeit at a low frequency when analyzed by an ultrasensitive method (0.009% to 26.9%), even before EGFR-TKI treatment [18]. It is hypothesized that these T790M-positive cells are protected from the effects of EGFR-TKI treatment and resistance acquired via the T790M mutation occurs frequently. Analysis using a highly sensitive method showed that the T790M mutation is present in 68% of cases of EGFR-TKI acquired resistance [19]. Osimertinib was developed based on the clear proof of concept that T790M was a potential target for therapeutic drug development and was successfully applied in clinical settings, even though it could not achieve a radical cure for NSCLC. Amplification of genes, such as MET and ERBB2 , transformation to small cell lung cancer, hepatocyte growth factor (HGF) overexpression, EMT transition, and PIK3CA mutation/reduced PTEN expression are other events that can occur after treatment with first- and second-generation EGFR-TKIs and with cancer progression, leading to the development of drug resistance [20]. In addition, Akt is frequently activated in tumors after treatment with first- and second-generation EGFR-TKIs [21]. However, none of the drugs targeting these mechanisms have so far been applied in clinical settings.
The mechanisms underlying resistance to osimertinib, a third-generation EGFR-TKI, have been investigated [22][23], and the presence of the EGFR C797S mutation in addition to the T790M mutation (T790M/C797S double-positive) has been proposed as one of the main mechanisms responsible for resistance acquisition in T790M mutation-positive lung cancer [24]. C797S reduces the affinity between osimertinib and the EGFR kinase domain and increases the relative affinity for ATP. EGFR-TKIs that can inhibit the kinase activity of EGFR in T790M/C797S double-positive lung cancer have also been developed, but their clinical usefulness is yet to be approved. In the case of T790M loss, several genetic alterations have been observed, including those related to transformation to small cell lung cancer, MET amplification, ERBB2 amplification/mutation, PIK3CA mutation, KRAS mutations, and oncogenic gene fusion involving RET , FGFR3 , and BRAF .
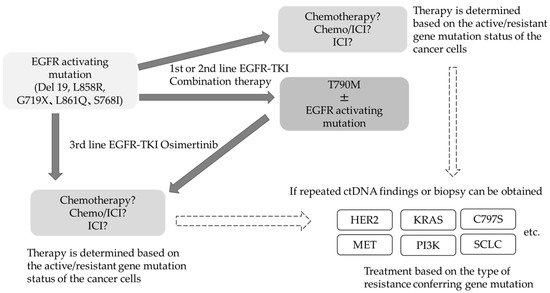
The frequency and spectrum of resistance mechanisms observed when osimertinib is used as first-line therapy in T790M-negative patients are different from those in T790M-positive patients after second-line therapy [23]. The incidence of drug resistance acquisition has been increasing and becoming more complex in nature. In addition to the drug resistance conferred by the C797S mutation in EGFR , amplification of the MET gene as a bypass pathway, alteration in RAS-RAF-MAPK pathway, ROS1 fusion gene, PIK3CA mutation, and transformation to small cell lung cancer are other mechanisms that impart drug resistance. Thus, the resistance mechanisms acquired after osimertinib treatment are rather complex, and therefore, it is challenging to treat osimertinib resistance using a single therapeutic approach. Even if specific resistance factors are identified, there are currently no insurance-approved therapies to target these factors. At present, the most notable therapeutic strategy for addressing acquired resistance is the combination of amivantamab, a bispecific antibody targeting EGFR and MET, and lazertinib, a third-generation EGFR-TKI. In phase I clinical trial conducted in EGFR mutation-positive patients with disease progression after osimertinib, the overall response rate for both drugs was 36% [25]. A subgroup analysis of MET-based resistant cases revealed good results, with a response rate of 75%. Although HER3 alterations are not known to mediate resistance to EGFR-TKIs, HER3-targeted patritumab deruxtecan was used in phase I clinical trials in EGFR-TKI resistant cases and produced a response rate of 39%, which is a good result, regardless of the background resistance mechanism [26]. With respect to treatment after failure of 3rd generation EGFR-TKIs, there is an ongoing debate as to whether to target a specific resistance-forming mechanism or to promote the development of drugs that are widely indicated regardless of the resistance-forming mechanism. The use of next-generation sequencing to confirm the details of these genetic mutations in daily practice remains uncommon for several reasons, affecting the pace of resistance factor identification. EGFR mutation-positive lung cancer is a classic example of monoclonal evolution in tumors. Even within an individual, the genomic landscape of individual tumors is heterogeneous, and the therapeutic agent of choice reveals the predominant subclones that are least susceptible to treatment over time [27]. Liquid biopsies have enabled the monitoring of genomic changes by analyzing circulating tumor (ct) DNA. Several clinical trials have reported that clearance of EGFR -activated mutations several weeks after the start of treatment is associated with good PFS, and liquid biopsy results have also become predictive markers for efficacy and residual lesions [28][29][30][31]. If the measurement of plasma ct DNA can be popularized in general practice, it would be possible to select treatments based on resistance status more widely ( Figure1 ).
References
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139.
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non—Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388.
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128.
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib Versus Crizotinib in Untreated ALK-positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838.
- Shaw, A.T.; Riely, G.J.; Bang, Y.J.; Kim, D.W.; Camidge, D.R.; Solomon, B.J.; Varella-Garcia, M.; Iafrate, A.J.; Shapiro, G.I.; Usari, T.; et al. Crizotinib in ROS1-Rearranged Advanced Non-Small-Cell Lung Cancer (NSCLC): Updated Results, Including Overall Survival, From PROFILE 1001. Ann. Oncol. 2019, 30, 1121–1126.
- Planchard, D.; Smit, E.F.; Groen, H.J.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib Plus Trametinib in Patients with Previously Untreated BRAF (V600E)-Mutant Metastatic Non-Small-Cell Lung Cancer: An Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1307–1316.
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282.
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943.
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640.
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50.
- Planchard, D.; Feng, P.-H.; Karaseva, N.; Kim, S.-W.; Kim, T.; Lee, C.; Poltoratskiy, A.; Yanagitani, N.; Marshall, R.; Huang, X.; et al. Osimertinib plus platinum–pemetrexed in newly diagnosed epidermal growth factor receptor mutation-positive advanced/metastatic non-small-cell lung cancer: Safety run-in results from the FLAURA2 study. ESMO Open 2021, 6, 100271.
- Kenmotsu, H.; Wakuda, K.; Mori, K.; Kato, T.; Sugawara, S.; Kirita, K.; Okamoto, I.; Azuma, K.; Nishino, K.; Teraoka, S.; et al. Primary Results of a Randomized Phase II Study of Osimertinib Plus Bevacizumab Versus Osimertinib Monotherapy for Un-treated Patients with Non-Squamous Non-Small Cell Lung Cancer Harboring EGFR Mutations: WJOG9717L Study. Ann. Oncol. 2021, 32, LBA44.
- Nguyen, K.-S.H.; Kobayashi, S.; Costa, D.B. Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non–Small-Cell Lung Cancers Dependent on the Epidermal Growth Factor Receptor Pathway. Clin. Lung Cancer 2009, 10, 281–289.
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26.
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors—Impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507.
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792.
- Aparicio, S.; Caldas, C. The Implications of Clonal Genome Evolution for Cancer Medicine. N. Engl. J. Med. 2013, 368, 842–851.
- Watanabe, M.; Kawaguchi, T.; Isa, S.-I.; Ando, M.; Tamiya, A.; Kubo, A.; Saka, H.; Takeo, S.; Adachi, H.; Tagawa, T.; et al. Ultra-Sensitive Detection of the Pretreatment EGFR T790M Mutation in Non—Small Cell Lung Cancer Patients with an EGFR-Activating Mutation Using Droplet Digital PCR. Clin. Cancer Res. 2015, 21, 3552–3560.
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of Lung Cancer Patientswith Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin. Cancer Res. 2011, 17, 1169–1180.
- Suda, K.; Mizuuchi, H.; Maehara, Y.; Mitsudomi, T. Acquired resistance mechanisms to tyrosine kinase inhibitors in lung cancer with activating epidermal growth factor receptor mutation—diversity, ductility, and destiny. Cancer Metastasis Rev. 2012, 31, 807–814.
- Jacobsen, K.; Bertran-Alamillo, J.; Molina, M.A.; Teixidó, C.; Karachaliou, N.; Pedersen, M.H.; Castellvi, J.; Garzón, M.; Servat, C.C.; Codony-Servat, J.; et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat. Commun. 2017, 8, 1–14.
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534.
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737.
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132.
- Bauml, J.; Cho, B.C.; Park, K.; Lee, K.H.; Cho, E.K.; Kim, D.-W.; Kim, S.-W.; Haura, E.B.; Sabari, J.K.; Sanborn, R.E.; et al. Amivantamab in combination with lazertinib for the treatment of osimertinib-relapsed, chemotherapy-naïve EGFR mutant (EGFRm) non-small cell lung cancer (NSCLC) and potential biomarkers for response. J. Clin. Oncol. 2021, 39, 9006.
- Janne, P.A.; Baik, C.S.; Su, W.C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated (EGFRm) Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2021, 39, 9007.
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548.
- Tsubata, Y.; Watanabe, K.; Saito, R.; Nakamura, A.; Yoshioka, H.; Morita, M.; Honda, R.; Kanaji, N.; Ohizumi, S.; Jingu, D.; et al. Osimertinib in poor performance status patients with T790M-positive advanced non-small-cell lung cancer after progression of first- and second-generation EGFR-TKI treatments (NEJ032B). Int. J. Clin. Oncol. 2021. Online ahead of print.
- Fukuhara, T.; Saito, H.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K.; et al. Evaluation of plasma EGFR mutation as an early predictor of response of erlotinib plus bevacizumab treatment in the NEJ026 study. EBioMedicine 2020, 57, 102861.
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive Detection of Response and Resistance in EGFR-Mutant Lung Cancer Using Quanti-tative Next-Generation Genotyping of Cell-Free Plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705.
- Ebert, E.B.F.; McCulloch, T.; Hansen, K.H.; Linnet, H.; Sorensen, B.; Meldgaard, P. Clearing of circulating tumour DNA predicts clinical response to osimertinib in EGFR mutated lung cancer patients. Lung Cancer 2020, 143, 67–72.

