Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, F. Dp71 Dystrophin Isoform. Encyclopedia. Available online: https://encyclopedia.pub/entry/16864 (accessed on 27 July 2026).
Wang F. Dp71 Dystrophin Isoform. Encyclopedia. Available at: https://encyclopedia.pub/entry/16864. Accessed July 27, 2026.
Wang, Faqi. "Dp71 Dystrophin Isoform" Encyclopedia, https://encyclopedia.pub/entry/16864 (accessed July 27, 2026).
Wang, F. (2021, December 07). Dp71 Dystrophin Isoform. In Encyclopedia. https://encyclopedia.pub/entry/16864
Wang, Faqi. "Dp71 Dystrophin Isoform." Encyclopedia. Web. 07 December, 2021.
Copy Citation
Dystrophin is a 427 kDa protein that stabilizes muscle cell membranes through interactions with the cytoskeleton and various membrane-associated proteins.
dystrophin
Dp71
Duchenne muscular dystrophy
hDMDdel52 mice
del52
1. Introduction
Dystrophin is a large, 427 kDa membrane-associated protein with a critical role in maintaining muscle membrane integrity. It has four main functional domains: an actin-binding domain (encoded by exons 1–8) at the N-terminus, a central rod domain (encoded by exons 8–64), a cysteine-rich domain (encoded by exons 64–70), and a C-terminal domain (encoded by exons 71–79) [1]. The specific function of each of these domains ultimately enables dystrophin to link the F-actin cytoskeleton in muscle fibers to the surrounding extracellular matrix, through a network of sarcolemmal glycoproteins (i.e., the dystrophin glycoprotein complex or DGC). This linkage provides mechanical reinforcement to the sarcolemma and protects it from stress during muscle contraction [2]. Mutations in the DMD gene that code for dystrophin lead to a group of disorders called the dystrophinopathies, of which the most prominent member is the fatal Duchenne muscular dystrophy (DMD). DMD is an X-linked recessive genetic disorder primarily characterized by progressive muscle degeneration and weakness [3]. About 1 in 3500–5000 males are born with DMD worldwide [4], and currently there is no cure for this disease apart from measures aimed at symptom management.
Expression of the full-length DMD transcript (Dp427) is independently regulated by three promoters. Each of these drives the expression of a version of Dp427 in different tissues, e.g., cortical neurons/hippocampus (B promoter) [5], cerebellar Purkinje cells/skeletal muscle (P promoter) [6], and skeletal/cardiac muscle (M promoter; primary full-length dystrophin) [5]. In addition to these Dp427 promoters, the DMD gene has at least four more internal promoters (at exons 30, 45, 56, and 63) that can produce shorter isoforms, each generating a dystrophin protein product named after their corresponding size, i.e., Dp260, Dp140, Dp116, and Dp71. Dp260 is expressed in the retina [7], Dp140 in the central nervous system and kidney [8], and Dp116 in Schwann cells [9]. Dp71 is expressed ubiquitously [10][11].
Even though the roles of full-length dystrophin have already been extensively studied, the physiological functions of its shorter isoforms are not well understood. Of particular interest would be Dp71, not only because it is expressed in muscle but also because previous works have implicated Dp71 in DMD pathogenesis [12][13]. Dp71 lacks both the N-terminal actin-binding and central rod domains of full-length dystrophin, rendering it unable to link muscle cell membranes to the cytoskeleton [14]. However, Dp71 retains the ability to bind proteins typically associated with the C-terminal end of Dp427, which could affect full-length dystrophin function. Indeed, one study showed that overexpression of human Dp71 in mice had a dominant negative effect on Dp427 and caused skeletal muscle degeneration [14]. In another study, Dp71 upregulation in the heart was associated with Purkinje fiber degeneration in a dystrophin-deficient dog model [13]. Clinical data from DMD patients revealed that mutations in the C-terminal domain, and in particular those affecting Dp71, were associated with decreased wheelchair use [15].
2. del52;WT Mice Have Significantly Elevated Dp71 Levels in the Heart but Not Skeletal Muscle, and Have Mostly Normal Skeletal Muscle Function
Western blotting with an anti-C-terminal dystrophin antibody revealed that del52;WT mice had significantly increased levels of Dp71 protein in the heart (p < 0.05) compared to wild-type mice (Figure 1A). Dp71 levels appeared elevated in the tibialis anterior of del52;WT mice but were non-significant. On the other hand, Dp427 levels were similar between del52;WT and wild-type mice. To determine the effect of increased Dp71 levels on skeletal muscle function, grip strength and rotarod tests were performed on wild-type, mdx, and del52;WT mice at 3, 6, 9, and 12 months. Forelimb grip strength was significantly reduced in del52;WT compared to wild-type mice at 6 months (p < 0.01), but not at 3, 9, or 12 months (Figure 1B), while mdx mice displayed significantly reduced forelimb grip strength versus wild-type only at 12 months (p < 0.05). No other significant differences between groups were observed within each age. A significant decline in forelimb grip strength was observed for wild-type mice at later ages (p < 0.01), which was not observed for mdx or del52;WT mice. As for total limb grip strength, wild-type and del52;WT mice had similar performance at all ages examined (Figure 1B). At 3 and 9 months, mdx mice had significantly reduced total limb grip strength than either wild-type or del52;WT mice (at least p < 0.05), while del52;WT mice showed a significant decline of total limb grip strength with age (3 months vs. 9 months, p < 0.01; 3 months vs. 12 months, p < 0.001). Wild-type mice exhibited a similar behavior only between 3 and 12 months (p < 0.001). No significant differences in rotarod test performance were observed between the three groups of mice at all ages examined (Figure 1C).
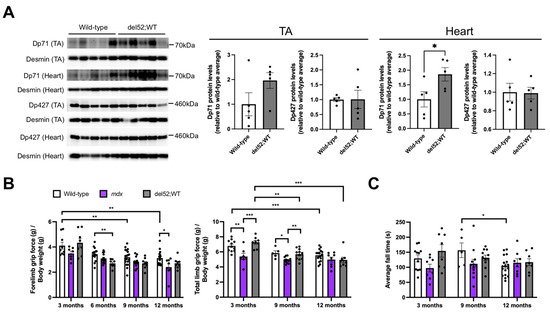
Figure 1. Dp71 overexpression and skeletal muscle function in del52;WT mice. (A) Western blotting with an antibody targeting the dystrophin C-terminal domain was done using total protein extracts from the tibialis anterior (TA) and the heart (12 μg each) of 6-month-old wild-type and del52;WT mice. Desmin was detected as a loading control. Quantification of Dp427 and Dp71 protein is on the right, with levels expressed relative to the average intensity of the corresponding wild-type samples for each tissue. Error bars: SEM, n = 5/group, * p < 0.05, unpaired two-tailed t-test. (B) Forelimb and total limb grip strength across age for wild-type, mdx, and del52;WT mice. Values are normalized to body weight. (C) Average fall times on the rotarod test across age. Dots represent individual mice. For (B,C), error bars: SEM, n = 5–19 (wild-type), 7–14 (mdx), 8–11 (del52;WT), * p < 0.05, ** p < 0.01, *** p < 0.001, one-way ANOVA with Tukey’s test.
3. Cardiac Function in del52;WT Mice Shows Signs of Impairment
To determine the consequences of elevated Dp71 levels in the heart of del52;WT mice, we performed cardiac phenotyping on wild-type, mdx, and del52;WT mice through electrocardiography, echocardiography, and invasive pressure-volume analysis. PR and QRS intervals were mostly similar between genotypes across age (Figure 2A). The corrected QT interval was significantly prolonged in del52;WT compared to wild-type or mdx mice at 9 months (p < 0.05) but was otherwise similar in other ages. While del52;WT mice also exhibited significantly slower heart rates than wild-type mice at 6 (p < 0.01) and 12 months (p < 0.05), mdx mice showed no detectible differences from wild-type mice in all parameters, save an increased QRS interval at 3 months (p < 0.001).
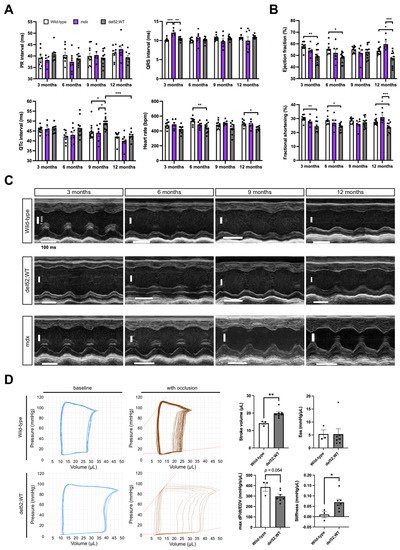
Figure 2. Cardiac function in del52;WT mice. (A) Electrocardiography results across age for wild-type, mdx, and del52;WT mice. (B) Ejection fraction and fractional shortening across age, as obtained through echocardiography. For (A,B), error bars: SEM, n = 8–12 (wild-type and del52;WT), 5–8 (mdx), * p < 0.05, ** p < 0.01, *** p < 0.001, one-way ANOVA with Tukey’s test. (C) Representative M-mode (parasternal long axis) echocardiography images of the left ventricle in the three groups of mice across age. (D) Representative pressure/volume loops at baseline and with occlusion for wild-type and del52;WT mice at 6 months are shown on the left. Quantification of selected parameters from the loops is shown on the right. Dots represent individual mice. Error bars: SEM, n = 3 (wild-type), 7 (del52;WT), * p < 0.05, ** p < 0.01, unpaired two-tailed t-test.
In terms of echocardiographic parameters, del52;WT mice had significantly reduced left ventricle (LV) ejection fraction and fractional shortening than wild-type mice at 3, 6, and 12 months (at least p < 0.05) or mdx mice at 12 months (p < 0.001) (Figure 2B); mdx mice had significantly increased LV ejection fraction and fractional shortening than wild-type mice at 12 months (p < 0.05). Cardiac structure was altered in del52;WT mice, which was especially evident at 3 months (Figure 2C). In particular, del52;WT mice had significantly thinner LV posterior walls than wild-type mice in at least one phase of the cardiac cycle (systole/diastole) across all ages examined (at least p < 0.05); this was also the case when compared with mdx mice at 6 months of age onward (at least p < 0.05) (Table 1). At 12 months, del52;WT mice had significantly increased systolic LV volumes versus wild-type mice (p < 0.01). Left atrium (LA) diameters were likewise significantly increased in del52;WT than wild-type mice, which was consistently observed up to 9 months of age (3, 6 months, p < 0.01; 9 months, p < 0.05). On the other hand, mdx mice showed significant LA dilation at 3 (p < 0.05) and 6 months (p < 0.01), as well as systolic LV posterior wall thinning at 6 months (p < 0.05) compared to wild-type mice. Moreover, mdx mice had decreased LV internal diameters and volumes (diastolic/systolic) at 9 and 12 months compared to both wild-type and del52;WT mice (p < 0.01).
Table 1. Summary of echocardiographic parameters in wild-type, mdx, and del52;WT mice across age.
| Parameter | 3 Months | 6 Months | 9 Months | 12 Months | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | mdx | del52;WT | WT | mdx | del52;WT | WT | mdx | del52;WT | WT | mdx | del52;WT | |
| Number | 8 | 4–5 | 12 | 8 | 5–6 | 11 | 10 | 3–6 | 11–12 | 12 | 6 | 10–11 |
| Heart rate | 443.0 (31.3) | 425.3 (32.8) | 426.6 (48.7) | 464.6 (32.7) | 450.5 (50.1) | 418.4 (36.8) b | 423.8 (45.6) | 528.7 (23.9) aa | 430.4 (38.9) cc | 427.1 (62.6) | 531.5 (63.4) aa | 393.3 (55.0) ccc |
| LVIDd | 3.9 (0.2) | 4.1 (0.2) | 4.2 (0.4) | 4.3 (0.4) | 4.0 (0.4) | 4.3 (0.3) | 4.3 (0.3) | 3.3 (0.4) aa | 4.5 (0.4) cc | 4.3 (0.2) | 3.5 (0.5) aa | 4.6 (0.4) cc |
| LVIDs | 2.8 (0.2) | 3.0 (0.2) | 3.2 (0.5) | 3.0 (0.3) | 2.9 (0.4) | 3.3 (0.3) | 3.1 (0.2) | 2.4 (0.3) aa | 3.3 (0.4) cc | 3.1 (0.2) | 2.4 (0.3) aa | 3.6 (0.4) cc |
| LVPWd | 0.6 (0.1) | 0.5 (0.2) | 0.5 (0.1) bb | 0.7 (0.05) | 0.7 (0.1) | 0.6 (0.1) bb,cc | 0.7 (0.1) | 0.8 (0.2) | 0.5 (0.1) b,cc | 0.6 (0.1) | 0.7 (0.1) | 0.5 (0.1) c |
| LVPWs | 1.0 (0.1) | 0.8 (0.2) | 0.7 (0.1) | 1.1 (0.1) | 0.9 (0.2) a | 0.8 (0.1) bb,c | 1.0 (0.1) | 1.1 (0.1) | 0.8 (0.1) bb,cc | 1.0 (0.2) | 1.1 (0.2) | 0.7 (0.2) bb,cc |
| LV vol s | 29.1 (4.1) | 34.1 (5.6) | 41.2 (15.4) | 36.3 (8.0) | 33.6 (12.1) | 44.1 (9.9) | 37.7 (4.6) | 20.9 (6.2) aa | 43.8 (11.7) cc | 38.4 (4.9) | 20.1 (7.2) aa | 54.0 (16.8) bb,cc |
| LV vol d | 68.1 (8.1) | 75.7 (7.1) | 79.8 (19.4) | 82.0 (16.4) | 68.9 (15.2) | 84.4 (15.6) | 84.7 (12.4) | 46.3 (12.9) aa | 92.2 (17.7) cc | 84.7 (7.0) | 51.6 (18.9) aa | 98.3 (18.4) cc |
| EF | 57.9 (4.0) | 54.3 (5.1) | 49.3 (7.2) bb | 55.7 (5.4) | 52.8 (7.6) | 48.2 (4.9) bb | 55.3 (3.4) | 52.1 (8.4) | 53.2 (5.9) | 53.9 (4.1) | 59.8 (5.7) a | 47.4 (7.1) bb,cc |
| FS | 30.1 (2.7) | 27.9 (3.3) | 24.8 (4.4) bb | 28.9 (3.7) | 27.0 (4.8) | 24.2 (2.9) bb | 28.6 (2.3) | 26.1 (4.8) | 27.4 (3.7) | 27.7 (2.7) | 31.0 (4.2) a | 23.8 (4.2) bb,cc |
| LA | 1.5 (52.4) | 1.6 (0.2) a | 1.6 (0.04) bb | 1.4 (0.1) | 1.7 (0.3) aa | 1.7 (0.1) bb | 1.4 (0.1) | 1.8 (0.7) | 1.9 (0.3) b | 1.6 (0.3) | 1.9 (0.5) | 1.9 (0.2) |
| IVRT | 16.9 (2.0) | 14.7 (2.1) | 16.5 (4.2) | 13.5 (1.0) | 14.1 (1.4) | 14.3 (1.8) | 13.4 (1.6) | 13.7 (2.4) | 13.3 (1.5) | 13.9 (2.0) | 12.6 (5.0) | 16.0 (2.7) |
| IVCT | 16.7 (56.6) | 13.9 (5.4) | 16.1 (5.5) | 13.5 (3.2) | 10.9 (2.7) | 12.5 (2.6) | 11.7 (3.3) | 10.5 (2.7) | 10.9 (2.9) | 12.3 (3.5) | 9.5 (2.4) | 13.8 (2.4) c |
| MV decel | 22.4 (4.9) | 18.3 (2.4) | 22.0 (5.6) | 17.7 (3.0) | 16.9 (5.4) | 24.5 (5.2) b,c | 19.8 (6.0) | 14.5 (7.2) | 22.9 (5.6) | 16.2 (4.6) | 16.5 (7.9) | 19.8 (3.5) |
| MV E | 663.5 (85.5) | 702.6 (70.6) | 681.9 (118.6) | 619.5 (65.7) | 713.8 (118.2) | 697.8 (76.1) | 610.8 (46.7) | 476.8 (196.5) | 727.9 (114.4) cc | 610.5 (36.8) | 541.5 (206.2) | 636.4 (103.0) |
| MV A | 476.0 (43.7) | 444.2 (65.1) | 438.5 (99.6) | 437.9 (92.8) | 513.8 (102.9) | 452.3 (98.1) | 423.2 (66.5) | 368.7 (179.0) | 458.2 (119.9) | 441.6 (102.3) | 412.9 (151.1) | 420.2 (178.2) |
| MV E/A | 1.4 (0.2) | 1.6 (0.2) | 1.6 (0.3) | 1.5 (0.2) | 1.4 (0.2) | 1.6 (0.4) | 1.5 (0.2) | 1.6 (1.2) | 1.7 (0.4) | 1.4 (0.3) | 1.6 (1.0) | 1.7 (0.4) |
| E’ | −24.6 (4.6) | −19.0 (2.8) | −23.6 (3.9) | −23.5 (5.5) | −25.8 (3.6) | −22.7 (3.3) | −23.1 (4.2) | −27.4 (4.5) | −21.8 (3.6) c | −19.1 (4.0) | −24.3 (9.5) | −18.0 (5.2) |
| A’ | −24.3 (5.3) | −26.9 (5.1) | −23.2 (1.8) | −22.9 (4.8) | −32.1 (9.3) a | −21.7 (3.6) cc | −21.1 (5.4) | −26.3 (5.2) | −23.8 (3.2) | −23.5 (5.0) | −27.5 (11.8) | −22.0 (6.9) |
| E’/A’ | 1.0 (0.2) | 0.7 (0.04) a | 1.0 (0.2) c | 1.1 (0.3) | 0.9 (0.3) | 1.1 (0.3) | 1.2 (0.4) | 1.1 (0.3) | 0.9 (0.2) | 0.9 (0.3) | 1.0 (0.7) | 0.9 (0.3) |
| MV E/E’ | −28.2 (8.8) | −37.5 (6.8) | −29.6 (6.7) | −27.9 (7.9) | −29.0 (6.4) | −31.3 (5.9) | −27.2 (5.3) | −17.5 (8.7) | −34.1 (6.3) b,cc | −33.1 (6.1) | −26.8 (16.0) | −38.1 (12.5) |
a—WT vs. mdx; b—WT vs. del52;WT; c—mdx vs. del52;WT; number of letters indicate significance, i.e., for a letter n, n p < 0.05, nn p < 0.01, nnn p < 0.001 (one-way ANOVA, post-hoc Tukey’s). Abbreviations: LVID—left ventricular interior diameter; LVPW—left ventricle posterior wall; EF—ejection fraction; FS—fractional shortening; LA—left atrium; IVRT—isovolumic relaxation time; IVCT—isovolumic contraction time; MV—mitral valve; d—diastole; s—systole, WT—wild-type.
Pressure–volume measurements at 6 months revealed that the pressure-volume loops of del52;WT mice were wider than those of wild-type mice (Figure 2D). Indeed, stroke volume was significantly elevated in del52;WT mice at this age (p < 0.01). Maximum dP/dt/EDV values were visibly reduced in del52;WT mice, but not statistically significant (p = 0.054). Evaluation of end-systolic and end-diastolic pressure–volume relationships through occlusion of the inferior vena cava showed that del52;WT hearts had no notable changes in contractility but had significantly stiffer LVs (p < 0.05), respectively.
4. del52;WT Mice Have Histologically Normal Skeletal and Cardiac Muscle
Finally, we stained tibialis anterior sections from wild-type and del52;WT mice with hematoxylin/eosin (HE) to assess the effects of Dp71 overexpression on overall skeletal muscle structure. For cardiac tissue sections, we performed immunofluorescent staining for collagen type I to study effects on fibrosis and cardiomyocyte size, two cellular phenotypes commonly affected in the dystrophic heart [16][17]. We did not notice any major differences between del52;WT and wild-type mice in these histological tests. HE staining revealed that del52;WT tibialis anterior muscles were histologically normal and indistinguishable from wild-type in terms of the absence of dystrophic landmarks such as fibrosis/necrosis, inflammatory infiltrates, and centrally-nucleated fibers (Figure 3A). Collagen staining was also similar between the two mice (Figure 3B), with no differences in the collagen type I-positive areas observed (Figure 3C). Measuring the minimal Feret’s diameter of individual cardiomyocytes in collagen type I-stained sections showed that del52;WT hearts had slightly smaller cardiomyocytes than those of wild-type mice, a difference that was statistically significant (p < 0.001) (Figure 3D,E).
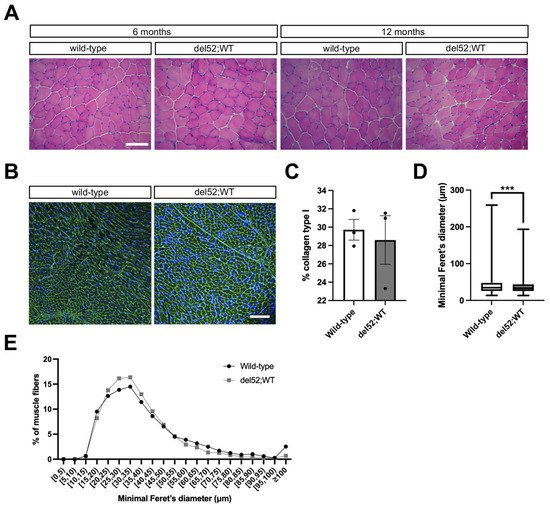
Figure 3. Histological analysis of skeletal and cardiac muscle in del52;WT mice. (A) Representative hematoxylin/eosin-stained images of tibialis anterior muscles in wild-type and del52;WT mice at 6 and 12 months. Scale bar: 100 μm. (B) Representative immunofluorescence images of collagen type I (green) and nuclei (blue) in the cardiac muscle of wild-type and del52;WT mice at 12 months. Scale bar: 100 μm. (C) Quantification of collagen type I area in cardiac muscle immunofluorescence images. Dots represent individual mice. Error bars: SEM, n = 3/group. (D) Quantification of the minimal Feret’s diameter of individual cardiomyocytes in cardiac muscle immunofluorescence images. Box: P25-P75, central line: median, whiskers: range. n = 3/group (average 1535 and 1951 fibers counted per replicate for wild-type and del52;WT, respectively), *** p < 0.001, unpaired two-tailed t-test. (E) Data in (D), but as a frequency distribution.
References
- Ervasti, J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 108–117.
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714.
- Manzur, A.Y.; Kinali, M.; Muntoni, F. Update on the management of Duchenne muscular dystrophy. Arch. Dis. Child. 2008, 93, 986–990.
- Hoffman, E.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928.
- Barnea, E.; Zuk, D.; Simantov, R.; Nudel, U.; Yaffe, D. Specificity of expression of the muscle and brain dystrophin gene promoters in muscle and brain cells. Neuron 1990, 5, 881–888.
- Gorecki, D.; Monaco, A.; Derry, J.M.J.; Walker, A.P.; Barnard, E.A.; Barnard, P.J. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum. Mol. Genet. 1992, 1, 505–510.
- D’Souza, V.N.; Man, N.T.; Morris, G.E.; Karges, W.; Pillers, D.-A.M.; Ray, P.N. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum. Mol. Genet. 1995, 4, 837–842.
- Lidov, H.G.W.; Selig, S.; Kunkel, L.M. Dp140: A novel 140 kDa CNS transcript from the dystrophin locus. Hum. Mol. Genet. 1995, 4, 329–335.
- Byers, T.J.; Lidov, H.G.W.; Kunkel, L.M. An alternative dystrophin transcript specific to peripheral nerve. Nat. Genet. 1993, 4, 77–81.
- Lederfein, D.; Levy, Z.; Augier, N.; Mornet, D.; Morris, G.; Fuchs, O.; Yaffe, D.; Nudel, U. A 71-kilodalton protein is a major product of the Duchenne muscular dystrophy gene in brain and other nonmuscle tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 5346–5350.
- Kawaguchi, T.; Niba, E.T.E.; Rani, A.Q.M.; Onishi, Y.; Koizumi, M.; Awano, H.; Matsumoto, M.; Nagai, M.; Yoshida, S.; Sakakibara, S.; et al. Detection of Dystrophin Dp71 in Human Skeletal Muscle Using an Automated Capillary Western Assay System. Int. J. Mol. Sci. 2018, 19, 1546.
- Greenberg, D.; Sunada, Y.; Campbell, K.; Yaffe, D.; Nudel, U. Exogenous Dp71 restores the levels of dystrophin associated proteins but does not alleviate muscle damage in mdx mice. Nat. Genet. 1994, 8, 340–344.
- Urasawa, N.; Wada, M.R.; Machida, N.; Yuasa, K.; Shimatsu, Y.; Wakao, Y.; Yuasa, S.; Sano, T.; Nonaka, I.; Nakamura, A.; et al. Selective Vacuolar Degeneration in Dystrophin-Deficient Canine Purkinje Fibers Despite Preservation of Dystrophin-Associated Proteins With Overexpression of Dp71. Circulation 2008, 117, 2437–2448.
- Leibovitz, S.; Meshorer, A.; Fridman, Y.; Wieneke, S.; Jockusch, H.; Yaffe, D.; Nudel, U. Exogenous Dp71 is a dominant negative competitor of dystrophin in skeletal muscle. Neuromuscul. Disord. 2002, 12, 836–844.
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry. J. Pers. Med. 2020, 10, 241.
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195.
- Tamiyakul, H.; Kemter, E.; Kösters, M.; Ebner, S.; Blutke, A.; Klymiuk, N.; Flenkenthaler, F.; Wolf, E.; Arnold, G.J.; Fröhlich, T. Progressive Proteome Changes in the Myocardium of a Pig Model for Duchenne Muscular Dystrophy. iScience 2020, 23.
More
Information
Subjects:
Allergy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
777
Revisions:
2 times
(View History)
Update Date:
08 Dec 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No