Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emilio Bucio | + 2933 word(s) | 2933 | 2021-11-04 04:18:12 | | | |
| 2 | Rita Xu | Meta information modification | 2933 | 2021-12-08 02:44:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bucio, E. Hydrogels Classification. Encyclopedia. Available online: https://encyclopedia.pub/entry/16859 (accessed on 02 August 2026).
Bucio E. Hydrogels Classification. Encyclopedia. Available at: https://encyclopedia.pub/entry/16859. Accessed August 02, 2026.
Bucio, Emilio. "Hydrogels Classification" Encyclopedia, https://encyclopedia.pub/entry/16859 (accessed August 02, 2026).
Bucio, E. (2021, December 07). Hydrogels Classification. In Encyclopedia. https://encyclopedia.pub/entry/16859
Bucio, Emilio. "Hydrogels Classification." Encyclopedia. Web. 07 December, 2021.
Copy Citation
Hydrogels are attractive biomaterials with favorable characteristics due to their water uptake capacity. However, hydrogel properties are determined by the cross-linking degree and nature, the tacticity, and the crystallinity of the polymer.
hydrogels
physical interactions
chemical interaction
1. Introduction
Novel biomaterials have been developed in the last decades. One of the most exciting for researchers is one of the first biomaterials, known as hydrogel [1]. The first hydrogel described arose in 1960, where the cross-linking network was based on polyhydroxyethylmethacrylate (pHEMA) [2][3][4]. A hydrogel is a tridimensional polymeric structure with swelling and collapse properties, flexibility, biodegradability, biocompatibility, and softness [5]. The ability of hydrogels to absorb water arises from hydrophilic functional groups attached to the polymeric backbone, while their resistance to dissolution arises from cross-links between network chains [6]. Variations in concentrations, structure, functionality of the monomer, and the cross-linker used in such gels can modify the structure [4]. Besides, these biocompatible materials have been widely used in the biomedical field due to their high ability to absorb drugs [7] and nanoparticles [8].
In hydrogel synthesis, monomers or polymers and an initiator are usually necessary. The latter will be responsible for the formation of monomeric free radicals that give rise to the growth of macromolecular chains. Monomers have unique properties that can form a macromolecular known as a polymer. In addition, a cross-linking agent is relevant because a characteristic of any hydrogel is its cross-linked structure, which is achieved through that agent [9][10]. The cohesion forces that allow the cross-linking of the polymer have a covalent character, and other forces such as electrostatic, hydrophobic, dipole–dipole interactions, or hydrogen bonds intervene [11][12][13]. Typically, hydrogels are classified into two categories: physically cross-linked hydrogels and chemically cross-linked hydrogels [14], which obtain different properties based on each synthesis.
In recent years, many efforts have been made on research approaches based on the synthesis of new hydrogels with better mechanical properties, which is one of these systems’ weak points, and provides them with a certain degree of “intelligence”. Since they are systems in an aqueous medium, it is necessary to consider the traditional variables such as temperature, concentration, pH, and ionic strength. Moreover, some hydrogels are capable of responding to external stimuli such as pH, temperature, electricity, light and biological molecules as enzymes during the swelling and shrinking process. Therefore, several studies have made it possible to improve mechanical, optical, or swelling behavior, adding another compound with hydrophobic properties to the hydrophilic monomer.
Hydrogels are composed of hydrophilic polymers, which are desirable materials in polymer science. They have different properties that make them potentially useful in a wide variety of applications, such as in biomedical applications, self-assembly, or catalysis [15]. Hydrophilic polymers might be considered as those polymers that contain polar functional groups such as hydroxyl (-OH), carboxyl (-COOH), and amino (-NH2) groups that make them soluble or swelled by water. A hydrophilic polymer that has received much attention is poly(vinyl alcohol) (PVA) because it has a great promise as a biological drug-delivery matrix [16] and is nontoxic. Similarly, hydrophilic polymers can be cross-linked through chemical bonds, leading to the formation of hydrogels, which are materials that have attracted particular attention in the biomedical field [17]. The research of hydrophilic polymers has been complex because the physical properties of solubility or swellability depend on different factors, such as the type of polymer, molecular weight, the ratio of polar groups, and degree of cross-linking. High molecular weight and a high degree of cross-linking will reduce the hydrophilicity of the molecule [18][19].
Hydrogels are attractive materials owing to their excellent features and properties. Besides, because of such a wide variety of response triggers, hydrogels can serve as sensors or actuators or can be utilized in controlled drug delivery systems, biosensors, tissue engineering scaffolds, and others [20], because of their biomimetic properties and multi functionalities [21].
2. Cross-Linking Strategies to Obtain Physical Hydrogels
Hydrogels can be cross-linked through physical or reversible networks, which hold them together by molecular entanglements or physicochemical interactions such as hydrogen bonds, hydrophobic interactions, charge condensation, or supramolecular chemistry. The interactions that occur in this type of hydrogels are weak. However, they are numerous and contribute to the presence of complex behaviors. Since the interactions depend significantly on external stimuli (pH, ionic strength, the composition of the solvent, or the temperature), they allow hydrogels to be highly versatile concerning the environment, unlike covalently bonded materials. Some of the gist hydrogels made up by the physicochemical are explained below.
2.1. Crystallization
Physical cross-linking of a polymer to form a hydrogel can also be achieved by crystallization through freeze–thaw cycles in homopolymeric systems or by forming stereocomplexes. The crystallization and degree of crystallinity determine the final properties of the resulting polymers. Polymeric crystallization can occur from dilute solutions or the molten state [22]. In the first case, crystallization occurs by evaporating the solvent, resulting in the appearance of single crystals based on a chain-folded model, which presents aligned chains [23]. This crystallization process includes crystal nucleation and crystal growth:
Crystal nucleation: The initial stage consists of the formation of tiny crystals or nuclei. It requires a certain degree of supersaturation, which increases the driving force for splitting the solution into a low and a high entropy region [24]. Several factors affect the crystal nucleation, such as temperature, volume, ionic strength, ionic species ratio, flow rate, and foreign particles [25].
Growth: It is characterized by the growth of the nuclei due to the alignment of molecular chain segments while part of the initial volume disappears. Polymers crystallize with chain-folded layers known as lamellae, which can be grouped forming spherical structures called spherulites. Polymers present crystalline phases when the lamellar chains are arranged in regular patterns and the amorphous phase when the lamellae are arranged irregularly. Generally, the polymers will crystallize from the melt in spherulitic structures [26]. The diameter of the spherulites is dependent on the nucleation sites, the molecular structure of the polymer, and the rate of cooling [27].
The crystallinity degree (%) measures the order in the molecular arrangement of polymers. It is calculated according to the following formula:
where, is the density of the completely crystalline polymer, represents the density of the completely amorphous polymer, and
is the density of the sample. The degree of crystallinity is dependent on the cooling rate and structure of the polymer [27]. It can range from a completely amorphous polymer (close to 0%) to a semicrystalline polymer (approximately 95%).
Freeze–thaw crystallization involves the formation of microcrystals in the structure. Examples of this type of cross-linking are xanthan [10][28] or PVA hydrogels [29][30]. The formation of resilient and resilient PVA gels is attributed to the formation of PVA crystallites that act as physical cross-linking sites in the network. The hydrogel properties depend on the concentration and molecular weight of the PVA, temperature, freezing time, and some freeze–thaw cycles [29]. It has been studied that by adding alginate to the PVA solution before subjecting it to the freeze–thaw process, the properties of the system can be modified (by increasing the concentration of alginate, the mechanical resistance increases, which causes a decrease in the drug release) [30].
Another form of crystallization is the formation of stereocomplexes [31]. Stereocomplexes are formed by stereoselective interactions between polymers with complementary stereoregular structures (the functional group is located only on one side of the monomer) that interact to form a system of properties different from those of the original constituents. It has been suggested that the forces involved in the formation of the complex are Van der Waals forces [32]. The formation and composition of the complexes between sterically complementary polymers are conditioned by medium composition or temperature [33].
2.2. Amphiphilic Copolymers
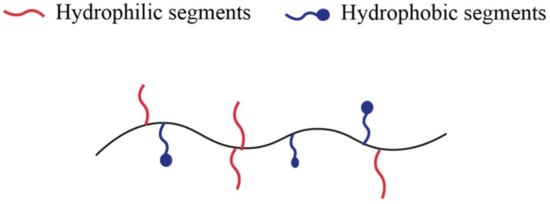
Amphiphilic hydrogels containing both hydrophilic and hydrophobic units represent one of the major polymeric biomaterials [34], as Figure 1 shows. In other words, amphiphilic copolymers can aggregate in water to form micelles and hydrogels in which the hydrophobic segments of the polymer self-assembly [35]. Copolymers typically form hydrogels with fragments of different nature or modified copolymers. The latter can be formed by a water-soluble polymer to which hydrophobic fragments have been attached or hydrophobic chains modified by water-soluble fragments [31].
Figure 1. Schematic illustration of amphiphilic polymer.
By the PEG biocompatibility and PLA biodegradability (or its copolymer with glycolic acid (PLGA)), the hydrogels formed by these copolymer blocks have been extensively investigated. Drug release can occur by passive diffusion and by degradation of the system. Multiple triblock polymer systems with hydrophobic segments in between have been proposed, for example, PEG-PLGA-PEG at low concentrations in water form micelles and high concentrations form thermoreversible hydrogels. The critical gelation concentration and the sol-gel transition temperature are highly dependent on the blocks’ molecular weights and composition. PEG-PLGA-PEG copolymers gel when there is a change from room temperature to 37 °C if the concentration is high enough. Cross-linking is thought to occur by hydrophobic interactions [36].
Another example is the copolymers of PLA and polyoxyethylene (PEO), PLA-PEO-PLA, formed by polymerization of L-lactide with PEG. It has been suggested that these hydrogels can retain hydrophilic drugs in the PEG phase and hydrophobic drugs in the PLA domains. Likewise, they can be applied by injection to administer specific proteins [37][38][39].
Copolymers containing fragments of different nature, such as composed of PEG and polybutylene terephthalate (a hydrophobic polyester) have also been investigated. Feijen et al. [40][41][42][43][44] studied that films or microspheres can be formed, and they tested the release of various proteins (lysozyme, bovine serum albumin). They also studied the hydrolytic degradation of polymers, concluding that as the percentage of polybutylene terephthalate increases, the degradation will be lower. Furthermore, modifying the water/polymer ratio during emulsification could control the release of proteins.
Polymers with hydrophobic domains can also crosslink in aqueous environments through reverse thermal gelling (sol-gel transition). Polymers or oligomers with this ability are known as gelling agents and are moderately hydrophobic [45][46][47]. Gelation occurs when a hydrophobic segment attaches to a hydrophilic polymer forming an amphiphilic polymer. Amphiphilic polymers are generally soluble in water at low temperatures; as the temperature increases, the hydrophobic domains add to minimize the hydrophobic surface area, reducing the amount of structured water around them [48]. Gelation temperature depends on the concentration of the polymer, the length of the hydrophobic block, and the chemical structure of the polymer. Some hydrophobic segments that can undergo reverse thermal gelation at temperatures close to physiological ones are PLGA, polypropylene oxide, poly(N-isopropyl acrylamide) (PNIPAAm), polypropylene fumarate, polycaprolactone, polyurethane, polyorganophosphazene [46].
Different polysaccharides (such as chitosan, dextran, pullulan, carboxymethyl curdlan) can also be modified by adding hydrophobic segments that self-assemble to form nanohydrogels. Sunamoto et al. [49][50][51] obtained cholesterol-modified pullulan nanoparticles. These 20–30 nm nanoparticles (nanohydrogels) can be loaded with different proteins such as α-chymotrypsin, bovine serum albumin, insulin [50]; or drugs such as adriamycin [52]. By covalently binding galactoside lactoside to pullulan, nanoparticles are obtained whose cellular target is RCA lectin (specific receptor for β-D-galactose).
Another example is chitosan glycol-modified with palmitoyl chains that assemble into unilamellar polymer vesicles in the presence of cholesterol [53]. These vesicles are biocompatible and hemocompatible and can encapsulate water-soluble drugs [54]. Chitosan has also been modified with various hydrophobic fragments to form pH or temperature-sensitive hydrogels; for example, D-L-lactic acid [55] and/or glycolic acid [56], polyacrylic acid (PAAc) [57], and PNIPAAm [58]. Other polymers such as carboxymethyl dextran have also been modified with poly(N-isopropyl acrylamide)-co-N,N-dimethyl acrylamide to form thermo-sensitive hydrogels [59]. Finally, another example along similar lines is the modification of carboxymethyl curdlan (polysaccharide with antitumor activity) with a sulfonylurea to form nanohydrogels by self-assembly [60].
2.3. Hydrogel Cross-Linking by Charge Interactions
Cross-linking (or de-crosslinking) can be achieved in situ by pH changes that cause ionization or protonation of ionic functional groups and cause gelation. Charge interactions can occur between a polymer and a small molecule or between two oppositely charged polymers to form a hydrogel. When a polyelectrolyte combines with a multivalent ion of opposite charge, a physical gel known as an ionotropic hydrogel is produced [46].
If two polyelectrolytes of opposite charges are mixed, they can gel or precipitate depending on their concentration, ionic strength, and pH of the solution; the product is called complex or polyionic. In pioneering work in the area, calcium alginate capsules were stabilized, coating them with an alginate-poly(L-lysine) coacervate complex [5].
Starch graft copolymers with neutralized acid monomers generate anionic starches that can gel through charge interactions. Prado et al. [61] reported the formation of a novel interpolyelectrolyte complex (IPEC) between cation-ized corn starch by introducing the 2-hydroxy-3 (N,N,N-trimethylammonium) propyl group and κ-type carrageenan as counterpolyanion. It should be noted that carrageenan is made up of galactose and/or anhydrogalactose units, sulfated or not, linked by alternating bonds. The κ-type consists of alternating galactose units with a sulfate group at carbon four and unsulfated anhydrogalactose units. Gao et al. developed a simple, nontoxic, water-based strategy to fabricate magnetic nanoparticles/hydrogels nanocomposites in which highly crystalline Fe3O4 nanoctahedra can be fabricated in situ within a negatively charged hydrogel matrix [62]. Besides, Katayama investigated electrostatic interactions in polyampholyte gels, which contain anions and cations in their skeleton [63]. They observed that they deflated at neutral pH (pH = 7), while at various pH values, both higher and lower, they swelled. The reason is that, at neutral pH, charges attract each other in such a way as to result in a deflation of the gel. Conversely, if one of the charges is neutralized and the other ionized, the gel swells. It is important to note that both van der Waals forces and hydrogen bonds cause collapse at low temperatures, while hydrophobic interactions cause the opposite effect. Electrostatic interactions can be attractive and repulsive, depending on the nature of the gel.
2.4. Interactions by Hydrogen Bonds
Hydrogen bonding interactions can be used to produce hydrogels in vitro by freeze–thaw cycles. An example is a project developed by You et al. in which they report a hydrogel with a hydrogen-bonding system consisting of weak hydrogen bonds between N,N-dimethylacrylamide (DMAA), and acrylic acid (AAc) and strong hydrogen bonds between 2-ureido-4 [H]-pyrimidinone units. The hydrogels have unique properties through optimization between the radii of the monomers and a balance of the interactions [64].
Yoshimura et al. prepared biodegradable hydrogels by a simple procedure: esterifying starch with succinic anhydride, using 4-dimethylaminopyridine as catalyst and dimethylsulfoxide or water as a solvent, followed by neutralization with NaOH, dialysis, and precipitation with methanol. These hydrogels were obtained in the absence of a cross-linker. The authors hypothesize that gelation occurred due to aggregation of polymer chains by regeneration of hydrogen bonds during dialysis [65].
2.5. Stereo-Complexing
Stereo-complexing refers to the interactions between polymeric chains, or small molecules, of the same chemical composition but different stereochemistry. Natural polymers can be cross-linked by stereo-complexing grafting. The grafting of L-lactide and D-lactide oligomers to dextran induces spontaneous gelation in water [46]. Dextran is a polysaccharide similar to amylopectin, consisting of highly branched glucose chains, whose predominant bond is α (1→6) with α (1→3) and α (1→4) branches. These hydrogels show excellent biocompatibility and biodegradability. They do not require organic solvents, chemical crosslinkers, or the formation of hydrophobic domains. However, the main drawback of stereo- complexation is the restricted polymer composition range that can be used; small changes in stoichiometry can weaken or eliminate the stereochemical interaction [46].
Some physical hydrogels have found interesting applications. For example, Mehyar et al. investigated two physical hydrogels (one based on hydrogen-bonded starch and the other based on calcium alginate, with ionic interactions) as possible substrates for antimicrobial agents. In these systems, diffusivity depended on the hydrogel-agent pair, being the highest with the following combinations: trisodium phosphate, starch hydrogel, and sodium acid chlorite-alginate hydrogel [66].
2.6. Protein Interactions
Cross-linking by protein interactions can be accomplished through the use of genetically engineered proteins or antigen–antibody interactions. By employing genetic engineering, a genetic code can be designed that originates peptide sequences with specific physicochemical properties, and even synthetic amino acids can be obtained [67]. Tirrell [68] and Cappello [69] were pioneers in this field.
Cappello et al. [70] created a high molecular weight protein-polymer based on a sequence of silk-like amino acids and elastin, in which the insoluble silk-like segments associate in sheets or strands linked by hydrogen bonds. A particular subset of these silk-elastin-type protein compositions, called ProLastins, gelled in physiological solution. The sol-gel transition can be controlled by modifying the temperature, the conditions of the solution, and the additives, which can prevent or promote the crystallization of the chain through hydrogen bonds.
Tirrell et al. [71] used recombinant DNA methods to create artificial proteins that undergo reversible gelation in response to changes in pH or temperature. Proteins are composed of terminal leucine zipper domains flanking a flexible central segment formed by a water-soluble polyelectrolyte. The formation of coiled aggregates of the terminal domains in near-neutral aqueous solutions and at room temperature triggers the formation of a three-dimensional polymeric network, in which the polyelectrolyte retains the solvent and prevents chain precipitation. Increasing the pH or temperature dissociates the terminal aggregates, causing the dissolution of the hydrogel. The mild pH and temperature conditions in which the hydrogel is formed suggest that these hydrogels could be applied in encapsulation or controlled drug and cell release.
Another cross-linking by protein interactions is that formed by antigen-antibody interactions. For example, Miyata et al. [72] proposed modifying a PAAm hydrogel to give it the ability to reversibly swell in a buffer solution in response to a specific antigen. The system was prepared by binding an antigen and the corresponding antibody to the polymer network; thus, antigen–antibody binding increases the cross-links of the PAAm hydrogel. The competitive binding of free antigen in the medium causes a change in the hydrogel volume due to the breaking of these non-covalent cross-links. Furthermore, it was shown that the hydrogel behaves with shape memory and that gradual changes in antigen concentration can induce pulsatile permeation through the lattice.
References
- Korah, L.; Anilkumar, G.; Thomas, S. Hydrogels, DNA, and RNA polypeptides for the preparation of biomaterials. In Fundamental Biomaterials: Polymers, 1st ed.; Thomas, S., Balakrishnan, P., Sreekala, M., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; pp. 85–104. ISBN 978-008-102-195-8.
- Lee, S.; Kwon, I.; Park, K. Hydrogels for delivery of bioactive agents: A historical perspective. Adv. Drug Deliv. Rev. 2013, 65, 17–20.
- Kopeček, J. Hydrogel biomaterials: A smart future? Biomaterials 2007, 28, 5185–5192.
- Yahia, L.; Chirani, N.; Yahia, L.; Gritsch, L.; Motta, F.; Chirani, S.; Fare, S. History and applications of hydrogels. J. Biomed. Sci. 2015, 4.
- Caló, E.; Khutoryanskiy, V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267.
- Ahmed, E. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121.
- Ayyala, D.; Blake, D.A.; John, V.T.; Ayyala, R.S. A glaucoma drainage device incorporating a slow-release drug delivery system for the management of fibrosis. In Biomaterials and Regenerative Medicine in Ophtalmology, 2nd ed.; Chirila, T.V., Harkin, D.G., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; pp. 349–367. ISBN 978-0-08-100147-9.
- Bustamante-Torres, M.; Romero-Fierro, D.; Arcentales-Vera, B.; Pardo, S.; Bucio, E. Interaction between filler and polymeric matrix in nanocomposites: Magnetic approach and applications. Polymers 2021, 13, 2998.
- Hoffman, A. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2002, 54, 3–12.
- Hoffman, A. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23.
- Ross-Murphy, S.; McEvoy, H. Fundamentals of hydrogels and gelation. Br. Polym. J. 1986, 18, 2–7.
- Anderson, J.; Brannon, J. Concentration dependence of the distribution coefficient for macromolecules in porous media. J. Polym. Sci. Polym. Phys. Ed. 1981, 19, 405–421.
- Janáček, J.; Stoy, A.; Stoy, V. Mechanical behavior of partially hydrolyzed preswollen polyacrylonitrile networks. J. Polym. Sci. Polym. Symp. 2007, 53, 299–312.
- Sikdar, P.; Uddin, M.; Dip, T.; Islam, S.; Hoque, M.; Dhar, A.; Wu, S. Recent advances in the synthesis of smart hydrogels. Mater. Adv. 2021, 2, 4532–4573.
- Schmidt, B. Hydrophilic polymers. Polymers 2019, 11, 693.
- Peppas, N.A.; Hoffman, A.S. Hydrogel. In Biomaterials Science: An Introduction to Materials in Medicine, 4th ed.; Wagner, W., Sakiyama-Elbert, S., Zhang, G., Yaszemski, M., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2020; pp. 153–166.
- Wang, C. Biodegradable polymer microparticles for genetic vaccine delivery. In Molecular Interfacial Phenomena of Polymers and Biopolymers, 1st ed.; Chen, P., Ed.; Woodhead Publishing: Sawston, UK, 2005; pp. 510–537. ISBN 9781855739284.
- Finch, C.A. Hydrophilic polymers. In Specialty Polymers; Dyson, R.W., Ed.; Springer: Boston, MA, USA, 1987; pp. 65–82. ISBN 978-1-4615-7894-9.
- Deng, F.; Luo, X.B.; Ding, L.; Luo, S. Application of nanomaterials and nanotechnology in the reutilization of metal ion from wastewater. In Nanomaterials for the Removal of Pollutants and Resource Reutilization, 1st ed.; Luo, D., Deng, F., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 149–178. ISBN 9780128148389.
- Peppas, N.; Slaughter, B.; Kanzelberger, M. Hydrogels. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier Science, BV: Amsterdam, The Netherlands, 2012; pp. 385–395. ISBN 978-0-08-087862-1.
- Liu, S.; Chen, X.; Zhang, Y. Hydrogels and hydrogel composites for 3D and 4D printing applications. In 3D and 4D Printing of Polymer Nanocomposite Materials; Sadasivuni, K.K., Deshmukh, K., Almaaded, M.A., Eds.; Elsevier Inc: Amsterdam, The Netherlands, 2020; pp. 427–465. ISBN 978-0-12-816805-9.
- Cyras, V.; D’Amico, D.; Manfredi, L. Crystallization behavior of polymer nanocomposites. In Crystallization in Multiphase Polymer Systems, 1st ed.; Thomas, S., Gowd, E.B., Arif, M., Kalarikkal, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 269–311. ISBN 978-0-12-809-453-2.
- Crawford, C.Q.; Quinn, B. Physiochemical properties and degradation. In Microplastic Pollutants; Crawford, C.Q., Quinn, B., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2017; pp. 57–100. ISBN 978-0-12-809406-8.
- Schulz, G. Protein crystallization. In Comprehensive Medicinal Chemistry II; Taylor, J., Triggle, D., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2007; pp. 433–446. ISBN 978-0-08-045044-5.
- Pan, H.; Jiang, S.; Zhang, T.; Tang, R. In situ solution study of calcium phosphate crystallization kinetics. Res. Methods Biominer. Sci. 2013, 532, 129–144.
- Chan, C.; Li, L. Direct observation of the growth of lamellae and spherulites by AFM. In Intrinsic Molecular Mobility and Toughness of Polymers II, 1st ed.; Henning Kausch, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–41. ISBN 978-3-540-31601-5.
- Balani, K.; Verma, V.; Agarwal, A.; Narayan, R. Physical, thermal, and mechanical propeerties of polymers. In Biosurfaces: A Materials Science and Engineering Perspective, 1st ed.; Balani, K., Verma, V., Agarwal, A., Narayan, R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 329–344. ISBN 978-111-895-062-3.
- Giannouli, P.; Morris, E. Cryogelation of xanthan. Food Hydrocoll. 2003, 17, 495–501.
- Yokoyama, F.; Masada, I.; Shimamura, K.; Ikawa, T.; Monobe, K. Morphology and structure of highly elastic poly (vinyl alcohol) hydrogel prepared by repeated freezing-and-melting. Colloid Polym. Sci. 1986, 264, 595–601.
- Takamura, A.; Ishii, F.; Hidaka, H. Drug release from poly (vinyl alcohol) gel prepared by freeze-thaw procedure. J. Control. Release 1992, 20, 21–27.
- Hennink, W.; van Nostrum, C. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 13–36.
- Slager, J.; Domb, A. Biopolymer stereocomplexes. Adv. Drug Deliv. Rev. 2003, 55, 549–583.
- Ravve, A. Physical properties and physical chemistry of polymers. In Principles of Polymer Chemistry, 3rd ed.; Ravve, A., Ed.; Springer: New York, NY, USA, 2012; pp. 17–67. ISBN 978-1-4614-2212-9.
- Chang, H.; Li, C.; Huang, R.; Su, R.; Qi, W.; He, Z. Amphiphilic hydrogels for biomedical applications. J. Mater. Chem. B 2019, 7, 2899–2910.
- Förster, S.; Antonietti, M. Amphiphilic block copolymers in structure-controlled nanomaterial hybrids. Adv. Mater. 1999, 10, 195–217.
- Jeong, B.; Bae, Y.; Kim, S. Thermoreversible gelation of PEG−PLGA−PEG triblock copolymer aqueous solutions. Macromolecules 1999, 32, 7064–7069.
- Rashkov, I.; Manolova, N.; Li, S.; Espartero, J.; Vert, M. Synthesis, characterization, and hydrolytic degradation of PLA/PEO/PLA triblock copolymers with short poly (l-lactic acid) chains. Macromolecules 1996, 29, 50–56.
- Li, S.; Rashkov, I.; Espartero, J.; Manolova, N.; Vert, M. Synthesis, characterization, and hydrolytic degradation of PLA/PEO/PLA triblock copolymers with long poly (l-lactic acid) blocks. Macromolecules 1996, 29, 57–62.
- Molina, I.; Li, S.; Martinez, M.; Vert, M. Protein release from physically crosslinked hydrogels of the PLA/PEO/PLA triblock copolymer-type. Biomaterials 2001, 22, 363–369.
- Bezemer, J.; Radersma, R.; Grijpma, D.; Dijkstra, P.; van Blitterswijk, C.; Feijen, J. Microspheres for protein delivery prepared from amphiphilic multiblock copolymers. 2. Modulation of release rate. J. Control. Release 2000, 67, 249–260.
- Bezemer, J.; Radersma, R.; Grijpma, D.; Dijkstra, P.; van Blitterswijk, C.; Feijen, J. Microspheres for protein delivery prepared from amphiphilic multiblock copolymers. 1. Influence of preparation techniques on particle characteristics and protein delivery. J. Control. Release 2000, 67, 233–248.
- Bezemer, J.; Grijpma, D.; Dijkstra, P.; van Blitterswijk, C.; Feijen, J. Control of protein delivery from amphiphilic poly (ether ester) multiblock copolymers by varying their water content using emulsification techniques. J. Control. Release 2000, 66, 307–320.
- Bezemer, J.; Radersma, R.; Grijpma, D.; Dijkstra, P.; Feijen, J.; van Blitterswijk, C. Zero-order release of lysozyme from poly (ethylene glycol)/poly (butylene terephthalate) matrices. J. Control. Release 2000, 64, 179–192.
- Bezemer, J.; Grijpma, D.; Dijkstra, P.; van Blitterswijk, C.; Feijen, J. A controlled release system for proteins based on poly (ether ester) block-copolymers: Polymer network characterization. J. Control. Release 1999, 62, 393–405.
- García, L.; Aguilar, M.R.; Román, J.S. Biodegradable hydrogels for controlled drug release. In Biomedical Applications of Hydrogels Handbook, 1st ed.; Ottenbrite, R., Park, K., Okano, T., Eds.; Springer: New York, NY, USA, 2010; pp. 147–155. ISBN 978-1-4419-5919-5.
- Hoare, T.; Kohane, D. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007.
- Frutos, G.; Prior-Cabanillas, A.; París, R.; Quijada-Garrido, I. A system novel controlled drug delivery based on pH-responsive hydrogels included in soft gelatin capsules. Acta Biomater. 2010, 6, 4650–4656.
- Guiseppi-Elie, A. Electroconductive hydrogels: Synthesis, characterization and biomedical applications. Biomaterials 2010, 31, 2701–2716.
- Akiyoshi, K.; Deguchi, S.; Tajima, H.; Nishikawa, T.; Sunamoto, J. Microscopic Structure and thermoresponsiveness of a hydrogel nanoparticle by self-assembly of a hydrophobized polysaccharide. Macromolecules 1997, 30, 857–861.
- Akiyoshi, K.; Kobayashi, S.; Shichibe, S.; Mix, D.; Baudys, M.; Wan Kim, S.; Sunamoto, J. Self-assembled hydrogel nanoparticle of cholesterol-bearing pullulan as a carrier of protein drugs: Complexation and stabilization of insulin. J. Control. Release 1998, 54, 313–320.
- Akiyoshi, K.; Deguchi, S.; Moriguchi, N.; Yamaguchi, S.; Sunamoto, J. Self-aggregates of hydrophobized polysaccharides in water. formation and characteristics of nanoparticles. Macromolecules 1993, 26, 3062–3068.
- Akiyoshi, K.; Taniguchi, I.; Fukui, H.; Sunamoto, J. Hydrogel nanoparticle formed by self-assembly of hydrophobized polysaccharide. Stabilization of adriamycin by complexation. Eur. J. Pharm. Biopharm. 1996, 42, 286–290.
- Uchegbu, I.; Schätzlein, A.; Tetley, L.; Gray, A.; Sludden, J.; Siddique, S.; Mosha, E. Polymeric chitosan-based vesicles for drug delivery. J. Pharm. Pharmacol. 2011, 50, 453–458.
- Sludden, J.; Uchegbu, I.; Schätzlein, A. The encapsulation of bleomycin within chitosan based polymeric vesicles does not alter its biodistribution. J. Pharm. Pharmacol. 2010, 52, 377–382.
- Qu, X.; Wirsén, A.; Albertsson, A. Structural change and swelling mechanism of ph-sensitive hydrogels based on chitosan andd, L-lactic acid. J. Appl. Polym. Sci. 1999, 74, 3186–3192.
- Qu, X.; Wirsén, A.; Albertsson, A. Novel Ph-sensitive chitosan hydrogels: Swelling behavior and states of water. Polymer 2000, 41, 4589–4598.
- Yazdani-Pedram, M.; Retuert, J.; Quijada, R. Hydrogels based on modified chitosan, 1. Synthesis and swelling behavior of poly (acrylic acid) grafted chitosan. Macromol. Chem. Phys. 2000, 201, 923–930.
- Kim, S.; Cho, S.; Lee, Y.; Kim, S. Thermo-and ph-responsive behaviors of graft copolymer and blend based on chitosan andn-isopropylacrylamide. J. Appl. Polym. Sci. 2000, 78, 1381–1391.
- Huh, K.; Hashi, J.; Ooya, T.; Yui, N. Synthesis and characterization of dextran grafted with poly (n-isopropylacrylamide-co-n,n-dimethyl-acrylamide). Macromol. Chem. Phys. 2000, 201, 613–619.
- Na, K.; Park, K.; Kim, S.; Bae, Y. Self-assembled hydrogel nanoparticles from curdlan derivatives: Characterization, anti-cancer drug release and interaction with a hepatoma cell line (Hepg2). J. Controlled Release 2000, 69, 225–236.
- Prado, H.; Matulewicz, M.; Bonelli, P.; Cukierman, A. Preparation and characterization of a novel starch-based interpolyelectrolyte complex as matrix for controlled drug release. Carbohydr. Res. 2009, 344, 1325–1331.
- Gao, Y.; Wei, Z.; Li, F.; Mao, Z.; Mei, Y.; Zrinyib, M.; Osada, Y. Synthesis of a morphology controllable Fe3O4 nanoparticle/hydrogel magnetic nanocomposite inspired by magnetotactic bacteria and its application in H2O2 detection. Green Chem. 2014, 16, 1255–1261.
- Katayama, S.; Hirokawa, Y.; Tanaka, T. Reentrant phase transition in acrylamide-derivative copolymer gels. Macromolecules 1984, 17, 2641–2643.
- You, Y.; Yang, J.; Zheng, Q.; Wu, N.; Lv, Z.; Jiang, Z. Ultra-stretchable hydrogels with hierarchical hydrogen bonds. Sci. Rep. 2020, 10, 1–8.
- Yoshimura, T.; Yoshimura, R.; Seki, C.; Fujioka, R. Synthesis and characterization of biodegradable hydrogels based on starch and succinic anhydride. Carbohydr. Polym. 2006, 64, 345–349.
- Mehyar, G.; Liu, Z.; Han, J. Dynamics of antimicrobial hydrogels in physiological saline. Carbohydr. Polym. 2008, 74, 92–98.
- Tirrell, J.; Tirrell, D. Synthesis of biopolymers: Proteins, polyesters, polysaccharides and polynucleotides. Curr. Opin. Solid State Mater. Sci. 1996, 1, 407–411.
- McGrath, K.; Fournier, M.; Mason, T.; Tirrell, D. Genetically directed syntheses of new polymeric materials. expression of artificial genes encoding proteins with repeating-(alagly) 3proglugly-elements. J. Am. Chem. Soc. 1992, 114, 727–733.
- Cappello, J.; Crissman, J.; Dorman, M.; Mikolajczak, M.; Textor, G.; Marquet, M.; Ferrari, F. Genetic engineering of structural protein polymers. Biotechnol. Prog. 1990, 6, 198–202.
- Cappello, J.; Crissman, J.; Crissman, M.; Ferrari, F.; Textor, G.; Wallis, O.; Whitledge, J.; Zhou, X.; Burman, D.; Aukerman, L.; et al. In-situ self-assembling protein polymer gel systems for administration, delivery, and release of drugs. J. Controlled Release 1998, 53, 105–117.
- Petka, W.; Harden, J.; Mcgrath, K.; Wirtz, D.; Tirrel, D. Reversible hydrogels from self-assembling artificial proteins. Science 1998, 281, 389–392.
- Miyata, T.; Asami, N.; Uragami, T. A Reversibly antigen-responsive hydrogel. Nature 1999, 399, 766–769.
More
Information
Subjects:
Biochemical Research Methods
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.4K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
08 Dec 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No