 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Salvatore Passarella | + 2719 word(s) | 2719 | 2021-11-29 04:14:52 | | | |
| 2 | Jessie Wu | Meta information modification | 2719 | 2021-12-08 03:24:02 | | |
Video Upload Options
The L-lactate (L-LAC)-mitochondria affair has its closure: that mitochondria can take up and metabolize L-LAC due to the presence of the mitochondrial L-lactate dehydrogenase is shown.
1. Introduction
Mitochondria play a key role in cell metabolism, and they govern a significant cross talk with the cytosol. This task is achieved by sharing metabolic pathways which rely on both cytosolic and mitochondrial enzymes. Mitochondria export metabolites/ATP for cytosol anaplerosis and import metabolite/ADP for final oxidation and ATP synthesis. Given that mitochondria are “close spaces” within the cytosol, they possess several translocators involved in cytosol communication with the inner mitochondrial compartments. However, as reported by Taylor [1] “the transport selectivities of many carriers remain unknown, and most have not been functionally investigated in mammalian cells”. A review describing “the multifaceted contributions of mitochondria to cellular metabolism” ignored or incompletely reported the transport and metabolism of certain metabolites in mitochondria, including L- and D-lactate and glutamine [2]. Papers describing the role of mitochondria in cancer do not take into consideration key transport processes, e.g., the transport into mitochondria of L-LAC which is the main product of cancer cell energy metabolism [3][4][5][6][7][8].
2. L-Lactate Transport in Mitochondria
2.1. The L-Lactate History
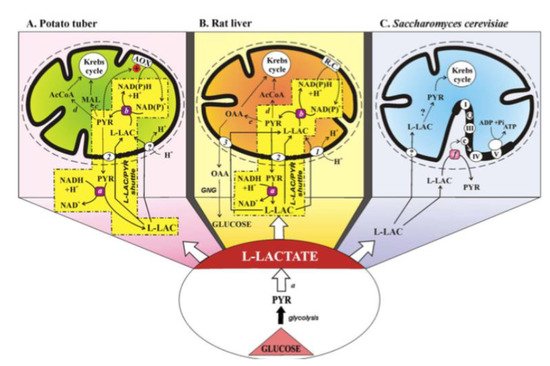
2.2 The L-Lactate Mitochondrial Metabolism Plays a Major Role in Neuronal Cells
2.3. The L-Lactate Mitochondrial Metabolism Plays a Major Role in Cancer Cells
References
- Taylor, E.B. Functional Properties of the Mitochondrial Carrier System. Trends Cell Biol. 2017, 27, 633–644.
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754.
- Adeva-Andany, M.; Lopez-Ojen, M.; Funcasta-Calderon, R.; Ameneiros-Rodriguez, E.; Donapetry-Garcia, C.; Vila-Altesor, M.; Rodriguez-Seijas, J. Comprehensive review on lactate metabolism in human health. Mitochondrion 2014, 17, 76–100.
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676.
- Lytovchenko, O.; Kunji, E.R.S. Expression and putative role of mitochondrial transport proteins in cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 641–654.
- Gladden, L.B. Lactate as a key metabolic intermediate in cancer. Ann. Transl. Med. 2019, 7, 210.
- Glancy, B.; Kane, D.A.; Kavazis, A.N.; Goodwin, M.L.; Willis, W.T.; Gladden, L.B. Mitochondrial lactate metabolism: History and implications for exercise and disease. J. Physiol. 2021, 599, 863–888.
- Cassim, S.; Vucetic, M.; Zdralevic, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119.
- Passarella, S.; de Bari, L.; Valenti, D.; Pizzuto, R.; Paventi, G.; Atlante, A. Mitochondria and L-lactate metabolism. FEBS Lett. 2008, 582, 3569–3576.
- de Bari, L.; Valenti, D.; Atlante, A.; Passarella, S. L-lactate generates hydrogen peroxide in purified rat liver mitochondria due to the putative L-lactate oxidase localized in the intermembrane space. FEBS Lett. 2010, 584, 2285–2290.
- Hashimoto, T.; Brooks, G.A. Mitochondrial lactate oxidation complex and an adaptive role for lactate production. Med. Sci. Sports Exerc. 2008, 40, 486–494.
- Paventi, G.; Lessard, C.; Bailey, J.L.; Passarella, S. In boar sperm capacitation L-lactate and succinate, but not pyruvate and citrate, contribute to the mitochondrial membrane potential increase as monitored via safranine O fluorescence. Biochem. Biophys. Res. Commun. 2015, 462, 257–262.
- Siesjo, B.K. Cell damage in the brain: A speculative synthesis. J. Cereb. Blood Flow Metab. 1981, 1, 155–185.
- Schurr, A.; Dong, W.Q.; Reid, K.H.; West, C.A.; Rigor, B.M. Lactic acidosis and recovery of neuronal function following cerebral hypoxia in vitro. Brain Res. 1988, 438, 311–314.
- Schurr, A.; West, C.A.; Rigor, B.M. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 1988, 240, 1326–1328.
- Izumi, Y.; Benz, A.M.; Zorumski, C.F.; Olney, J.W. Effects of lactate and pyruvate on glucose deprivation in rat hippocampal slices. Neuroreport 1994, 5, 617–620.
- Larrabee, M.G. Partitioning of CO2 production between glucose and lactate in excised sympathetic ganglia, with implications for brain. J. Neurochem. 1996, 67, 1726–1734.
- Larrabee, M.G. Lactate metabolism and its effects on glucose metabolism in an excised neural tissue. J. Neurochem. 1995, 64, 1734–1741.
- Schurr, A.; Payne, R.S.; Miller, J.J.; Rigor, B.M. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: An in vitro study. Brain Res. 1997, 744, 105–111.
- Schurr, A.; Payne, R.S.; Miller, J.J.; Rigor, B.M. Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: Further in vitro validation. J. Neurochem. 1997, 69, 423–426.
- Hu, Y.; Wilson, G.S. A temporary local energy pool coupled to neuronal activity: Fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J. Neurochem. 1997, 69, 1484–1490.
- Schurr, A.; Miller, J.J.; Payne, R.S.; Rigor, B.M. An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J. Neurosci. 1999, 19, 34–39.
- Schurr, A.; Payne, R.S.; Miller, J.J.; Tseng, M.T.; Rigor, B.M. Blockade of lactate transport exacerbates delayed neuronal damage in a rat model of cerebral ischemia. Brain Res. 2001, 895, 268–272.
- Smith, D.; Pernet, A.; Hallett, W.A.; Bingham, E.; Marsden, P.K.; Amiel, S.A. Lactate: A preferred fuel for human brain metabolism in vivo. J. Cereb. Blood Flow Metab. 2003, 23, 658–664.
- Dalsgaard, M.; Volianitis, S.; Yoshiga, C.C.; Dawson, E.A.; Secher, N.H. Cerebral metabolism during upper and lower body exercise. J. Appl. Physiol. 2004, 97, 1733–1739.
- Schurr, A. Lactate: The ultimate cerebral oxidative energy substrate? J. Cereb. Blood Flow Metab. 2006, 26, 142–152.
- Schurr, A.; Payne, R. Lactate, not pyruvate, is neuronal aerobic glycolysis end product: An in vitro electrophysiological study. Neuroscience 2007, 147, 613–619.
- Holmes, B.E.; Holmes, E.G. Contributions to the Study of Brain Metabolism: Carbohydrate Metabolism. (Preliminary Paper.). Biochem. J. 1925, 19, 492–499.
- Holmes, B.E.; Holmes, E.G. Contributions to the Study of Brain Metabolism: Carbohydrate Metabolism. Biochem. J. 1925, 19, 836–839.
- Holmes, E.G.; Holmes, B.E. Contributions to the Study of Brain Metabolism: Carbohydrate Metabolism Relationship of Glycogen and Lactic Acid. Biochem. J. 1926, 20, 1196–1203.
- Holmes, B.E.; Holmes, E.G. Contributions to the Study of Brain Metabolism. IV: Carbohydrate Metabolism of the Brain Tissue of Depancreatised Cats. Biochem. J. 1927, 21, 412–418.
- Holmes, E.G.; Ashford, C.A. Lactic acid oxidation in brain with reference to the “Meyerhof cycle”. Biochem. J. 1930, 24, 1119.
- Holmes, E. The metabolism of brain and nerve. Annu. Rev. Biochem. 1932, 1, 487–506.
- Ashford, C.A.; Holmes, E.G. Contributions to the study of brain metabolism: Role of phosphates in lactic acid production. Biochem. J. 1929, 23, 748–759.
- Ashford, C.A.; Holmes, E.G. Further observations on the oxidation of lactic acid by brain tissue. Biochem. J. 1931, 25, 2028.
- Quastel, J.H.; Wheatley, A.H. Oxidations by the brain. Biochem. J. 1932, 26, 725–744.
- Krebs, H.A.; Johnson, W.A. The role of citric acid in intermediate metabolism in animal tissues. FEBS Lett. 1980, 117 (Suppl. 1), K1–K10.
- Krebs, H.A.; Johnson, W.A. Metabolism of ketonic acids in animal tissues. Biochem. J. 1937, 31, 645–660.
- Krebs, H.A.; Johnson, W.A. Acetopyruvic acid (αγ-diketovaleric acid) as an intermediate metabolite in animal tissues. Biochem. J. 1937, 31, 772–779.
- O’Brien, J.; Kla, K.M.; Hopkins, I.B.; Malecki, E.A.; McKenna, M.C. Kinetic parameters and lactate dehydrogenase isozyme activities support possible lactate utilization by neurons. Neurochem. Res. 2007, 32, 597–607.
- Wyss, M.T.; Jolivet, R.; Buck, A.; Magistretti, P.J.; Weber, B. In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 2011, 31, 7477–7485.
- Schurr, A. Lactate: A major and crucial player in normal function of both muscle and brain. J. Physiol. 2008, 586, 2665.
- Larsen, T.S.; Rasmussen, P.; Overgaard, M.; Secher, N.H.; Nielsen, H.B. Non-selective beta-adrenergic blockade prevents reduction of the cerebral metabolic ratio during exhaustive exercise in humans. J. Physiol. 2008, 586, 2807–2815.
- Schurr, A. and Gozal, E. Aerobic production and utilization of lactate satisfy increased energy demands upon neuronal activation in hippocampal slices and provide neuroprotection against oxidative stress. Front. Pharmacol. 2012, 2, 96.
- Schurr, A. Glycolysis Paradigm Shift Dictates a Reevaluation of Glucose and Oxygen Metabolic Rates of Activated Neural Tissue. Front. Neurosci. 2018, 12, 700.
- Atlante, A.; de Bari, L.; Bobba, A.; Marra, E.; Passarella, S. Transport and metabolism of L-lactate occur in mitochondria from cerebellar granule cells and are modified in cells undergoing low potassium dependent apoptosis. Biochim. et Biophys. Acta 2007, 1767, 1285–1299.
- Kennedy, K.M.; Dewhirst, M.W. Tumor metabolism of lactate: The influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 2010, 6, 127–148.
- De Bari, L.; Chieppa, G.; Marra, E.; Passarella, S. L-lactate metabolism can occur in normal and cancer prostate cells via the novel mitochondrial L-lactate dehydrogenase. Int. J. Oncol. 2010, 37, 1607–1620.
- Hussien, R.; Brooks, G.A. Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol Genom. 2011, 43, 255–264.
- Passarella, S.; Atlante, A.; Valenti, D.; de Bari, L. The role of mitochondrial transport in energy metabolism. Mitochondrion 2003, 2, 319–343.
- Pizzuto, R.; Paventi, G.; Porcile, C.; Sarnataro, D.; Daniele, A.; Passarella, S. l-Lactate metabolism in HEP G2 cell mitochondria due to the l-lactate dehydrogenase determines the occurrence of the lactate/pyruvate shuttle and the appearance of oxaloacetate, malate and citrate outside mitochondria. Biochim. et Biophys. Acta 2012, 1817, 1679–1690.
- Passarella, S.; Schurr, A. l-Lactate Transport and Metabolism in Mitochondria of Hep G2 Cells-The Cori Cycle Revisited. Front. Oncol. 2018, 8, 120.
- de Bari, L.; Atlante, A. Including the mitochondrial metabolism of L-lactate in cancer metabolic reprogramming. Cell. Mol. Life Sci. CMLS 2018, 75, 2763–2776.


