 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexis Kalergis | + 2383 word(s) | 2383 | 2021-11-16 04:25:28 | | | |
| 2 | Camila Xu | Meta information modification | 2383 | 2021-12-08 01:29:07 | | |
Video Upload Options
Epigenetic mechanisms, such as DNA methylation, histone modifications, and non-coding RNAs, are known regulators of gene expression and genomic stability in cell growth, development, and differentiation. Since epigenetic mechanisms can regulate several immune system elements, epigenetic alterations have been found in several autoimmune diseases.
1. Introduction
Autoimmune diseases are characterized by the breakdown of self-tolerance and the presence of self-reactive immune cells [1]. Among them, we will focus on the most frequent diseases, including Rheumatoid Arthritis (RA), Systemic Lupus Erythematosus (SLE), and others, such as Multiple Sclerosis (MS), Sjogren’s Syndrome (SS), and Psoriasis. Although the etiology of autoimmune diseases is associated with a complex genetic susceptibility, it is clear that genes are not the only factors contributing to disease [2]. Indeed, when evaluating the development of autoimmune diseases in genetically identical monozygotic twins, environmental factors can contribute substantially to developing autoimmune disorders [3]. Epigenetic mechanisms can be influenced by environmental factors and be heritable, including microRNAs, post-transcriptional modifications (PTMs) of histones, and DNA methylation [4]. These mechanisms alter chromatin architecture, control the accessibility to transcriptional regulatory factors, and regulate gene transcription rates.
DNA methylation and gene expression profiles are unique and specific to each cell type, including immune cell subsets [5]. Methylation profiling on peripheral blood mononuclear cells (PBMCs) samples may be masking cell-type-specific epigenetic signatures, such as marked differences in methylome studies between lymphoid and myeloid cells [6]. Therefore, methylation and transcriptomic studies carried on purified cell subsets will provide accurate results.
Methylome and transcriptome studies in female PBMCs demonstrated that monocytes and B cells display distinct and unique gene clustering while CD4+ and CD8+ T cells patterns gather together [6] ( Figure 1 ). Differentially methylated genes in each immune cell subsets are located mainly downstream promoter-CGIs, exons, and introns [6]. However, several methylation differences could be found up or downstream shores and shelves [6]. Contrary to expected, the methylation status of promoter-bearing CpG sites displays fewer frequencies than total CpG sites [6]. Differential methylation genes (DMG) specific to cell subsets are not so frequent in CGIs. Additionally, the authors found that these non-CGIs’ differentially methylated regions (DMR) are located in enhancer elements and may regulate immune cell homeostasis functions [6]. These observations indicate that gene body methylation status plays a relevant role in cell-type-specific immune transcription and function.
Naive T cells usually displayed 5mC in transcriptional regulatory regions, such as promoter sites of cytokine locus overlapping with conserved non-coding sequences resulting in Th gene silencing ( Figure 1 ). Non-coding DNA sequences may contain binding sites for transcription factors or other molecules involved in transcription regulation [7]. IFNG, IL4, and IL17 genes displayed 5hmC modifications, specifically in conserved non-coding sequences (CNS) and promoter regions of purified Th1, Th2, and Th17 T cells [8]. The CNS6 enhancer sequence at the IFNG gene is most hydroxymethylated in Th1 cells and hypermethylated in the other Th subsets. Similarly, CNS2 and IL17a promoters of the IL17 locus are highly hydroxymethylated in the Th17 subset but hypermethylated in different T cell subsets [8] ( Figure 1 ). Thus, 5mC and 5hmC found at lineage-cytokine genes strictly link to their expression in each Th subsets and highlight that active DNA demethylation is crucial for immune regulation in Th-lineage development. Naive CD4+ T cells expressed high levels of Tet demethylases, but after TCR engagement, most Tet members are down-regulated [9]. However, Tet2 remains highly expressed in all Th subsets suggesting a broad role in Th differentiation. Furthermore, Th1 cells displayed recruitment of Tet2 to 5hmC-enriched CNS-6 and promoter regions of the IFNG locus where the presence of the T-bet transcription factor would be essential [9]. Similarly, Tet2 together with RORγt achieves DNA demethylation in the IL17 locus. However, Th2-related genes are not as much targeted by Tet2 as Th1 or Th17. In contrast, as observed in CD4+ Th cells, during CD8+ T cells differentiation, Tet2 is much more linked to cell fate (effector/memory) than profile features or cytokine expression [8]. Tet2/3 are also associated with Pro-B to Pre-B transition or thymic T cell development as reviewed in Li et al. 2021; however, being outside the scope of the manuscript is not included [9].
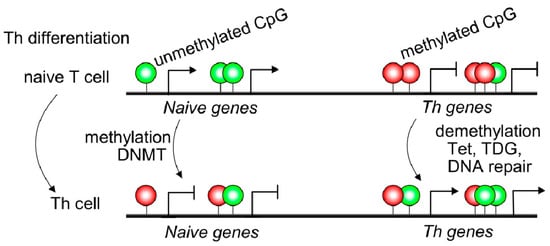
2. Dysregulated Epigenetic Modifications in Autoimmune Diseases
Alterations in DNA methylation are observed in the MHC region of CD14+ monocytes from RA patients, associated with altered gene regulation and an increased RA risk [10]. Besides, other authors have reported hypermethylation of IL10 [11] and IL6 [12] promoter regions in PBMCs from RA patients. Additionally, hypomethylation of the promoter of IL6 and ERα has been associated with increased expression in RA patients [13][14]. These alterations may be linked to increased IL-6 expression and Th17 cell proliferation [12]. Several hypomethylated genes have been detected in RA patients, such as GALNT9 in B and T cells [15]. Moreover, IL-6R, CAPN8, DPP4, CD74, CCR6, and several HOX genes in fibroblast-like synoviocytes have DNA methylation changes and accordingly showed a dysregulated expression ( Figure 2 ) [16]. Interestingly, there are different interactions between DNA methylations and miRNAs affecting gene regulation in an integrated way in RA patients [16].
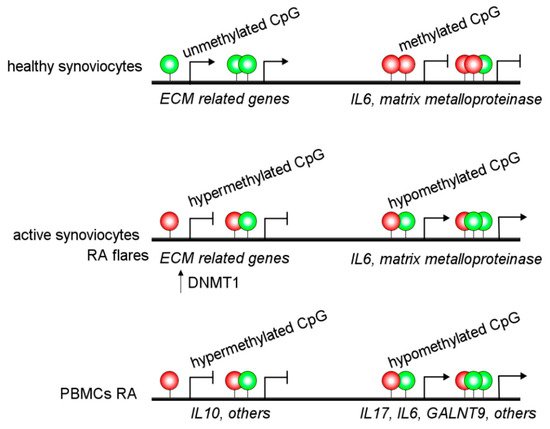
Recently, the epigenetic landscape was evaluated in RA fibroblast-like synoviocytes, which adopt an aggressive phenotype in RA patients. They studied histone modifications, chromatin structure, RNA expression, and DNA methylation and detected epigenetic changes associated with active enhancers, promoters, and specific transcription factor binding motifs [17]. Interestingly, this study reports a new way to identify unexpected RA-specific targets relevant to the development of novel therapeutic agents by considering the complexity of the epigenomic landscape [17].
SLE is mainly driven by B cells; however, several reports also link lupus immunopathogenesis with T cells, dendritic cells, and monocytes [18][19][20][21]. Furthermore, DNA methylation studies have also linked T cells to lupus pathogenesis [22][23]. Modifying DNA methylation of T cells during polyclonal proliferation with the DNMT inhibitors, 5-azacitidine (5Aza) and procainamide, may drive aberrant pro-inflammatory genes transcription and loss of tolerance [24]. The administration of activated and demethylated T cells into mice develops a Lupus-like disease, including anti-dsDNA production and glomerular immune complex deposition [24]. However, a different outcome was observed in a T cell-targeted 5Aza approach where demethylation occurs only in T cells [25]. MRL lpr lupus mice treated with nanolipogels loaded with 5Aza and tagged with anti-CD4, or -CD8 monoclonal antibodies ameliorated skin rash, proteinuria glomerular damage is reduced, and the inflammatory infiltration is decreased [25]. Surprisingly, authors found that targeting CD4+ T cells with this nanolipogel loaded with 5Aza, Foxp3 + Tregs displayed a marked expansion in spleen cervical lymph nodes. The authors also showed that the 5Aza treatment favors Foxp3 expression by inhibiting methylation in humans and mice treated CD4+ T cells. When nanolipogels were directed against CD8+ T cells, double-negative T cell subsets were reduced highly, suggesting a link between these two T cell populations [25]. In experimental models, absolute numbers of T and B cells, plasma cells, germinal center B cells, IFN-γ producing T cells, and effector/memory T cells were increased in the absence of Tet2 and Tet3 demethylases on B cells [26]. Furthermore, Tet2 and Tet3 deficiency leads to anti-dsDNA, -histone, and sm/RNP autoantibodies development, leading to renal immune-complexes deposition, which are significant features of lupus-like symptoms [26]. Indeed, when authors depleted CD4+ T cells or deleted H2-Ab1 (MHCII) gene, which prevents T-B cooperation, plasma cell numbers and T cell aberrant activation was decreased, suggesting that lymphocyte interaction is crucial in autoimmune initiation. In this work, the authors highlight the role of CD86 dysregulation on B cells and the subsequent T cell aberrant activation upon Tet2 and Tet3 deficiency in lupus-like disease [26]. Remarkably, the authors concluded that the lack of Tet2 and Tet3 conditions unleashes CD86 expression during continuous self-antigen exposure [26].
Psoriasis is linked to aberrant crosstalk between dendritic cells, T cells, and keratinocytes to produce multiple inflammatory cytokines and growth factors [27][28][29][30][31]. Strikingly, although extensive studies over immune skin cells have been done, regulation of the immune response by DNA methylation in psoriasis has barely been addressed. Interestingly, purified blood CD4+ cells from discordant monozygotic twins, one healthy and one affected with psoriasis, display a highly similar DNA methylation landscape [32]. However, minor differences in DNA methylation and expression exist in CD4+ T cells that gather genes, such as IL13 and TNFSF11, among others [32]. In addition, gene methylation profiles of psoriasis patients indicate that genes belonging to the IL17 signaling pathway, Staphylococcus aureus infection, interferons, and immune cells migration displayed abnormal methylation, including IRF7, IL7R, and CXCL1 [33]. Interestingly, the imiquimod induced psoriasis model in knocking down Tet2 mice displayed decreased skin lesions with a reduced expression of biomarkers genes, such as S100A7, IL7R, and IRF7. These data suggest that deficient methylation/demethylation homeostasis may contribute to disease risk.
3. Female and X-Linked DNA Methylation
In contrast, ERβ may display anti-inflammatory functions [34]. Interestingly, lower levels of ERβ may be found in lupus T cells [35]. Similarly, Crohn’s disease patients may also display a decrease in ERβ expression in blood T cells [36]. Although these data suggest that downregulation of ERβ may be linked to a pro-inflammatory condition, the correlation between expression and methylation has not yet been studied.
Biological markers are used to determine a normal situation, pathological processes, or the result of therapeutic interventions [37]. Thus, determining molecular markers in target tissue within the context of autoimmune diseases allows an in-depth understanding of the pathogenesis and the identification of new early diagnosis points and possible novel therapeutic targets. In addition, immune-related genes and inflammation pathways are under epigenetic-mediated regulation [1]. Accordingly, understanding the involvement of DNA methylation and histones modification in the pathogenesis of autoimmunity could provide a patient-specific drug-response prediction.
The phenotype heterogeneity and overlap within autoimmune diseases are some of the main aspects that make diagnosis difficult. However, early treatment of the disease can delay the onset of detrimental symptoms [38]. Therefore, this makes prompt intervention and accurate diagnosis critical for the patient’s progression. Although we have detailed multiple epigenetic alterations associated with autoimmune diseases throughout this review, most of these hallmarks have not yet been studied as biomarkers that allow early diagnosis. In addition, some epigenetic changes have been associated with disease progression. For example, low methylation of CYP2E1 and DUSP22 promoters have been associated with disease activity and could be used as a RA disease activity biomarker [39].
Plasma circulating miRNAs are ideal biomarkers for early autoimmune disease diagnosis and monitoring progression because they are stable and non-invasively detected in fluids. Importantly, several miRNAs (MiR-24, miR-26a, and miR-125a-5p) were reported to be increased in plasma from RA patients and thus, have been suggested as possible non-invasive biomarkers [40]. Notably, miR-24 and miR-125a-5p increase were specific for RA disease, and its level was reduced in SLE and osteoarthritis patients [40]. Moreover, five miRNAs (miR-103a-3p, miR-155-5p, miR-200a-3p, miR-210-3p, and miR-146a-5p) were suggested as potential Type 1 Diabetes (T1D) biomarkers as they were dysregulated in recently-diagnosed T1D patients [41].
4. Advantages and Disadvantages of Epigenetic Therapy in Autoimmunity
There are alterations in the epigenetic landscape that are shared by different autoimmune diseases [42]. Nevertheless, the altered expression of particular genes may help diagnose and determine new therapies among specific autoimmune disorders. Along these lines, recent work reported that a group of autoimmune diseases (RA, SLE, GD and, SSc) share the hypomethylation of IFN-related genes in CD4+ T cells and could be used as a signature for various autoimmune disorders [42]. Accordingly, aberrant type I IFN function has been implicated in several of the mentioned autoimmune diseases [42]. On the other hand, as discussed in the article, many genes and enzymes have been targeted as potential therapies for autoimmune diseases. Currently, preclinical and clinical trials have been made to test their security and efficiency. For instance, inhibitors of HDAC are widely used in medicine, and ITF2357 (givinostat) is administrated to children with an anti-inflammatory purpose for treating systemic-onset juvenile idiopathic arthritis [43]. This HDAC inhibitor has also been tested in animal models of autoimmune diseases, such as RA with promising results [43]. However, it is essential to consider that there is still a lack of information regarding the contribution of epigenetics to immune and non-immune responses [44].
Another example is using HDAC6 inhibitor as a treatment for SLE and inflammatory bowel disease in rodent models, showing anti-inflammatory effects via CKD-506. Nevertheless, the mechanism regarding inflammatory and non-inflammatory response and the cells involved remains unknown [45].
On the other hand, targets already described may serve in some ethnic groups but not all. For example, in RA disease, the upregulation of miR-499 rs3746444 increased risk, particularly in Caucasians [46]. Therefore, studies must correlate target genes with different population characteristics regarding ethnic groups, sex, age, etc.
Finally, it is crucial to consider that aside from epigenetics-targeted therapies, some regular treatment for autoimmune diseases may also lead to epigenetic profiles similar to healthy controls, such as methotrexate used for RA treatment. For example, it has been shown that methotrexate reduces methylation in the FOXP3 gene, restoring the Treg function by increasing FoxP3 and CTLA4 expression [47], in contrast to anti-TNFα therapy which has not been associated with DNA hypomethylation restoration in RA patients [48]. Moreover, dietary changes and microbiota alterations lead to changes in epigenetic (local and systemically). Therefore, intervention strategies (pre and probiotics) could be suitable for modifying epigenetic alterations [49]. Besides, several environmental factors, such as smoking which reduces DNA methylation, could increase the epigenetic risk [50].
References
- Zhang, P.; Lu, Q. Genetic and epigenetic influences on the loss of tolerance in autoimmunity. Cell. Mol. Immunol. 2018, 15, 575–585.
- Floreani, A.; Leung, P.S.C.; Gershwin, M.E. Environmental Basis of Autoimmunity. Clin. Rev. Allergy Immunol. 2016, 50, 287–300.
- Ballestar, E. Epigenetics lessons from twins: Prospects for autoimmune disease. Clin. Rev. Allergy Immunol. 2010, 39, 30–41.
- Liberman, N.; Wang, S.Y.; Greer, E.L. Transgenerational epigenetic inheritance: From phenomena to molecular mechanisms. Curr. Opin. Neurobiol. 2019, 59, 189–206.
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481.
- Mamrut, S.; Avidan, N.; Staun-Ram, E.; Ginzburg, E.; Truffault, F.; Berrih-Aknin, S.; Miller, A. Integrative analysis of methylome and transcriptome in human blood identifies extensive sex- and immune cell-specific differentially methylated regions. Epigenetics 2015, 10, 943–957.
- Wittkopp, P.J.; Kalay, G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2011, 13, 59–69.
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626.
- Li, J.; Li, L.; Sun, X.; Deng, T.; Huang, G.; Li, X.; Xie, Z.; Zhou, Z. Role of Tet2 in Regulating Adaptive and Innate Immunity. Front. Cell Dev. Biol. 2021, 9, 665897.
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M.; et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013, 31, 142–147.
- Fu, L.-H.; Ma, C.-L.; Cong, B.; Li, S.-J.; Chen, H.-Y.; Zhang, J.-G. Hypomethylation of proximal CpG motif of interleukin-10 promoter regulates its expression in human rheumatoid arthritis. Acta Pharmacol. Sin. 2011, 32, 1373–1380.
- Ishida, K.; Kobayashi, T.; Ito, S.; Komatsu, Y.; Yokoyama, T.; Okada, M.; Abe, A.; Murasawa, A.; Yoshie, H. Interleukin-6 gene promoter methylation in rheumatoid arthritis and chronic periodontitis. J. Periodontol. 2012, 83, 917–925.
- Liu, H.-W.; Lin, H.-L.; Yen, J.-H.; Tsai, W.-C.; Chiou, S.-S.; Chang, J.-G.; Ou, T.-T.; Wu, C.-C.; Chao, N.-C. Demethylation within the proximal promoter region of human estrogen receptor alpha gene correlates with its enhanced expression: Implications for female bias in lupus. Mol. Immunol. 2014, 61, 28–37.
- Nile, C.J.; Read, R.C.; Akil, M.; Duff, G.W.; Wilson, A.G. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008, 58, 2686–2693.
- Glossop, J.R.; Emes, R.D.; Nixon, N.B.; Haworth, K.E.; Packham, J.C. Genome-wide DNA methylation profiling in rheumatoid arthritis identifies disease-associated methylation changes that are distinct to individual T-and B-lymphocyte populations. Epigenetics 2014, 9, 1228–1237.
- de la Rica, L.; Urquiza, J.M.; Gómez-Cabrero, D.; Islam, A.B.M.M.K.; López-Bigas, N.; Tegnér, J.; Toes, R.E.M.; Ballestar, E. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J. Autoimmun. 2013, 41, 6–16.
- Ai, R.; Laragione, T.; Hammaker, D.; Boyle, D.L.; Wildberg, A.; Maeshima, K.; Palescandolo, E.; Krishna, V.; Pocalyko, D.; Whitaker, J.W. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat. Commun. 2018, 9, 1921.
- Herrada, A.A.; Llanos, C.; Mackern-Oberti, J.P.; Carreño, L.J.; Henriquez, C.; Gómez, R.S.; Gutierrez, M.A.; Anegon, I.; Jacobelli, S.H.; Kalergis, A.M. Haem oxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology 2012, 136, 414–424.
- Mackern-Oberti, J.P.; Llanos, C.; Carreño, L.J.; Riquelme, S.A.; Jacobelli, S.H.; Anegon, I.; Kalergis, A.M. Carbon monoxide exposure improves immune function in lupus-prone mice. Immunology 2013, 140, 123–132.
- Funes, S.C.; Ríos, M.; Gómez-Santander, F.; Fernández-Fierro, A.; Altamirano-Lagos, M.J.; Rivera-Perez, D.; Pulgar-Sepúlveda, R.; Jara, E.L.; Rebolledo-Zelada, D.; Villarroel, A.; et al. Tolerogenic dendritic cell transfer ameliorates systemic lupus erythematosus in mice. Immunology 2019, 158, 322–339.
- Mackern-Oberti, J.P.; Obreque, J.; Méndez, G.P.; Llanos, C.; Kalergis, A.M. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 182, 1–13.
- Moulton, V.R.; Tsokos, G.C. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J. Clin. Investig. 2015, 125, 2220–2227.
- Jeffries, M.; Dozmorov, M.; Tang, Y.; Merrill, J.T.; Wren, J.D.; Sawalha, A.H. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics 2011, 6, 593–601.
- Quddus, J.; Johnson, K.J.; Gavalchin, J.; Amento, E.P.; Chrisp, C.E.; Yung, R.L.; Richardson, B.C. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J. Clin. Investig. 1993, 92, 38–53.
- Li, H.; Tsokos, M.G.; Bickerton, S.; Sharabi, A.; Li, Y.; Moulton, V.R.; Kong, P.; Fahmy, T.M.; Tsokos, G.C. Precision DNA demethylation ameliorates disease in lupus-prone mice. JCI Insight 2018, 3, e120880.
- Tanaka, S.; Ise, W.; Inoue, T.; Ito, A.; Ono, C.; Shima, Y.; Sakakibara, S.; Nakayama, M.; Fujii, K.; Miura, I.; et al. Tet2 and Tet3 in B cells are required to repress CD86 and prevent autoimmunity. Nat. Immunol. 2020, 21, 950–961.
- Elder, J.T.; Bruce, A.T.; Gudjonsson, J.E.; Johnston, A.; Stuart, P.E.; Tejasvi, T.; Voorhees, J.J.; Abecasis, G.R.; Nair, R.P. Molecular dissection of psoriasis: Integrating genetics and biology. J. Investig. Dermatol. 2010, 130, 1213–1226.
- Christophers, E. Psoriasis--epidemiology and clinical spectrum. Clin. Exp. Dermatol. 2001, 26, 314–320.
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143.
- Cheuk, S.; Wiken, M.; Blomqvist, L.; Nylen, S.; Talme, T.; Stahle, M.; Eidsmo, L. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J. Immunol. 2014, 192, 3111–3120.
- Moreno-Sosa, T.; Sánchez, M.B.; Pietrobon, E.O.; Fernandez-Muñoz, J.M.; Zoppino, F.C.M.; Neira, F.J.; Germanó, M.J.; Cargnelutti, D.E.; Innocenti, A.C.; Jahn, G.A.; et al. Desmoglein-4 Deficiency Exacerbates Psoriasiform Dermatitis in Rats While Psoriasis Patients Displayed a Decreased Gene Expression of DSG4. Front. Immunol. 2021, 12, 708.
- Gervin, K.; Vigeland, M.D.; Mattingsdal, M.; Hammerø, M.; Nygård, H.; Olsen, A.O.; Brandt, I.; Harris, J.R.; Undlien, D.E.; Lyle, R. DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: Identification of epigenetically dysregulated genes. PLoS Genet. 2012, 8, e1002454.
- Wang, X.; Liu, X.; Liu, N.; Chen, H. Prediction of crucial epigenetically-associated, differentially expressed genes by integrated bioinformatics analysis and the identification of S100A9 as a novel biomarker in psoriasis. Int. J. Mol. Med. 2020, 45, 93–102.
- Li, J.; McMurray, R.W. Effects of estrogen receptor subtype-selective agonists on autoimmune disease in lupus-prone NZB/NZW F1 mouse model. Clin. Immunol. 2007, 123, 219–226.
- Maselli, A.; Conti, F.; Alessandri, C.; Colasanti, T.; Barbati, C.; Vomero, M.; Ciarlo, L.; Patrizio, M.; Spinelli, F.R.; Ortona, E.; et al. Low expression of estrogen receptor beta in T lymphocytes and high serum levels of anti-estrogen receptor alpha antibodies impact disease activity in female patients with systemic lupus erythematosus. Biol. Sex Differ. 2016, 7, 016–0057.
- Pierdominici, M.; Maselli, A.; Varano, B.; Barbati, C.; Cesaro, P.; Spada, C.; Zullo, A.; Lorenzetti, R.; Rosati, M.; Rainaldi, G.; et al. Linking estrogen receptor beta expression with inflammatory bowel disease activity. Oncotarget 2015, 6, 40443–40451.
- Aronson, J.K.; Ferner, R.E. Biomarkers—A general review. Curr. Protoc. Pharmacol. 2017, 76, 9–23.
- Wu, H.; Liao, J.; Li, Q.; Yang, M.; Zhao, M.; Lu, Q. Epigenetics as biomarkers in autoimmune diseases. Clin. Immunol. 2018, 196, 34–39.
- Mok, A.; Rhead, B.; Holingue, C.; Shao, X.; Quach, H.L.; Quach, D.; Sinclair, E.; Graf, J.; Imboden, J.; Link, T. Hypomethylation of CYP 2E1 and DUSP 22 Promoters Associated With Disease Activity and Erosive Disease Among Rheumatoid Arthritis Patients. Arthritis Rheumatol. 2018, 70, 528–536.
- Murata, K.; Furu, M.; Yoshitomi, H.; Ishikawa, M.; Shibuya, H.; Hashimoto, M.; Imura, Y.; Fujii, T.; Ito, H.; Mimori, T. Comprehensive microRNA analysis identifies miR-24 and miR-125a-5p as plasma biomarkers for rheumatoid arthritis. PLoS ONE 2013, 8, e69118.
- Assmann, T.S.; Recamonde-Mendoza, M.; Puñales, M.; Tschiedel, B.; Canani, L.H.; Crispim, D. MicroRNA expression profile in plasma from type 1 diabetic patients: Case-control study and bioinformatic analysis. Diabetes Res. Clin. Pract. 2018, 141, 35–46.
- Chen, S.; Pu, W.; Guo, S.; Jin, L.; He, D.; Wang, J. Genome-Wide DNA Methylation Profiles Reveal Common Epigenetic Patterns of Interferon-Related Genes in Multiple Autoimmune Diseases. Front. Genet. 2019, 10, 223.
- Joosten, L.A.; Leoni, F.; Meghji, S.; Mascagni, P. Inhibition of HDAC activity by ITF2357 ameliorates joint inflammation and prevents cartilage and bone destruction in experimental arthritis. Mol. Med. 2011, 17, 391–396.
- Ibáñez-Cabellos, J.S.; Seco-Cervera, M.; Osca-Verdegal, R.; Pallardó, F.V.; García-Giménez, J.L. Epigenetic regulation in the pathogenesis of Sjögren Syndrome and Rheumatoid Arthritis. Front. Genet. 2019, 10, 1104.
- Park, J.K.; Jang, Y.J.; Oh, B.R.; Shin, J.; Bae, D.; Ha, N.; il Choi, Y.; Youn, G.S.; Park, J.; Lee, E.Y. Therapeutic potential of CKD-506, a novel selective histone deacetylase 6 inhibitor, in a murine model of rheumatoid arthritis. Arthritis Res. Ther. 2020, 22, 176.
- Liu, F.; Liang, Y.; Zhao, Y.; Chen, L.; Wang, X.; Zhang, C. Meta-analysis of association of microRNAs genetic variants with susceptibility to rheumatoid arthritis and systemic lupus erythematosus. Medicine 2021, 100, e25689.
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Amjadi, P.; Green, P.; Read, J.E.; Brennan, F.; Gregory, B.; Williams, R.O. Methotrexate restores regulatory T cell function through demethylation of the FoxP3 upstream enhancer in patients with rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 1182–1192.
- Liu, C.-C.; Fang, T.-J.; Ou, T.-T.; Wu, C.-C.; Li, R.-N.; Lin, Y.-C.; Lin, C.-H.; Tsai, W.-C.; Liu, H.-W.; Yen, J.-H. Global DNA methylation, DNMT1, and MBD2 in patients with rheumatoid arthritis. Immunol. Lett. 2011, 135, 96–99.
- Wu, J.; Zhao, Y.; Wang, X.; Kong, L.; Johnston, L.J.; Lu, L.; Ma, X. Dietary nutrients shape gut microbes and intestinal mucosa via epigenetic modifications. Crit. Rev. Food Sci. Nutr. 2020, 1–15.
- Tsaprouni, L.G.; Yang, T.-P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Viñuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9, 1382–1396.



