Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristina Nocella | + 2587 word(s) | 2587 | 2021-11-18 07:07:07 | | | |
| 2 | Camila Xu | + 313 word(s) | 2900 | 2021-12-07 07:55:28 | | | | |
| 3 | Camila Xu | + 313 word(s) | 2900 | 2021-12-07 07:56:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nocella, C. Antiphospholipid Syndrome (APS). Encyclopedia. Available online: https://encyclopedia.pub/entry/16806 (accessed on 23 July 2026).
Nocella C. Antiphospholipid Syndrome (APS). Encyclopedia. Available at: https://encyclopedia.pub/entry/16806. Accessed July 23, 2026.
Nocella, Cristina. "Antiphospholipid Syndrome (APS)" Encyclopedia, https://encyclopedia.pub/entry/16806 (accessed July 23, 2026).
Nocella, C. (2021, December 06). Antiphospholipid Syndrome (APS). In Encyclopedia. https://encyclopedia.pub/entry/16806
Nocella, Cristina. "Antiphospholipid Syndrome (APS)." Encyclopedia. Web. 06 December, 2021.
Copy Citation
Antiphospholipid syndrome (APS) is considered an autoimmune, thrombo-inflammatory disease characterized by vascular thrombosis in the setting of one or more antiphospholipid antibodies (aPLs) such as lupus anticoagulant (AL), anticardiolipin antibodies (aCL) and anti-β2-glycoprotein1 antibodies (aβ2GPI).
oxidative stress
thrombosis
antiphospholipid syndrome
antioxidant treatment
1. Introduction
In the early 1980s, the term antiphospholipid syndrome (APS) was coined to describe an autoimmune, multisystemic disorder characterized clinically by autoantibody-induced thrombophilia [1]. Today, APS is considered an autoimmune, thrombo-inflammatory disease characterized by vascular thrombosis in the setting of one or more antiphospholipid antibodies (aPLs) such as lupus anticoagulant (AL), anticardiolipin antibodies (aCL), and anti-β2-glycoprotein1 antibodies (aβ2GPI) [2]. Beyond thrombosis, APS regularly manifests with other morbid features including thrombocytopenia, cardiac dysfunction [3], accelerated atherosclerosis, nephropathy, movement disorders, and cognitive decline [4][5]. This heterogeneous clinical presentation reflects the complex pathogenesis of APS, reinforcing the need for a deeper knowledge of mechanisms of aPL formation and of thrombotic complications, to allow a better-tailored, integrated, multidisciplinary approach.
It is known that the pathogenesis of APS consists of two phases: “the first hit and second hit”. According to this theory, the “first hit” is represented by the presence of circulating aPLs that destroy the integrity of the endothelium inducing a procoagulant phenotype. Nevertheless, aPLs alone are not enough to cause thrombosis, which takes place only in the presence of triggering factors (the “second hit”), which is usually represented by smoking, acute infections, oxidative stress, or inflammation [6].
2. Mechanisms of Atherothrombosis in APS: The Role of Oxidative Stress
The diagnosis of APS requires the concomitant presence of vascular thrombosis and/or pregnancy morbidity [7], in addition to persistent positivity to at least one of the aPL among LA, aCL, and aβ2GPI. However, some patients may present non criteria antibodies or unusual clinical manifestations [8][9]. Venous thromboembolism is the most common clinical presentation of the syndrome whereas arterial thrombosis is less frequent and mainly affects younger adults. The clinical spectrum of arterial thrombosis may extend from asymptomatic small ischemic lesions to fully ischemic stroke [10]. An observational study in 1000 APS patients from 13 European countries, that were followed prospectively for 10 years, showed that thrombotic events appeared in 16.6% during the first 5 years and 14.4% during the second 5 years. The most common events reported were strokes, transient ischaemic attacks, deep vein thrombosis, and pulmonary embolism [11].
It’s now well established that oxidative stress plays a major role in atherogenesis [12][13][14]. Oxidative stress is defined by an imbalance between reactive oxygen species (ROS) production and impaired detoxification by antioxidant enzymatic and nonenzymatic systems [15][16][17]. This imbalance characterizes several cardiovascular diseases (CVD) in which ROS are important mediators of endothelial damage leading to vascular inflammation and progression of the atherosclerotic plaque.
Several mechanisms have been proposed as promoters of oxidative stress in APS patients (Figure 1).
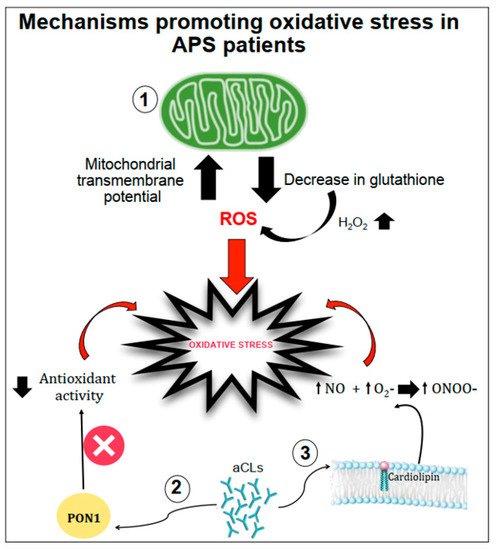
Figure 1. Schematic representation of mechanisms promoting oxidative stress in APS patients. Oxidative stress can be favoured by (1) the increase in the mitochondrial transmembrane potential and the decrease in intracellular glutathione contents; (2) the interactions between anticardiolipin antibodies (aCL) and the paraoxonase-1 (PON1) limiting its antioxidant properties; (3) aCL induction of nitric oxide (NO) and superoxide (O2−) production with increased levels of peroxynitrite (ONOO−) a pro-oxidant molecule.
Gergely et al. [18] verified, in patients with systemic lupus erythematosus, the hypothesis that the mitochondrial transmembrane potential and production of reactive oxygen intermediates (ROIs) mediate the imbalance of apoptosis which may significantly contribute to inflammation. In these patients, they found that mitochondrial transmembrane potential and ROI production were elevated compared to healthy subjects. Moreover, intracellular glutathione contents were diminished, and H2O2, a precursor of ROIs, increased mitochondrial transmembrane potential and caused apoptosis [18].
Another mechanism of oxidative stress can be related to the interactions between aCL antibodies and antioxidant enzymes in plasma, such as the paraoxonase-1 (PON1), which is an antioxidant enzyme linked to HDLs that prevents LDL oxidation. Indeed, in patients positive for aCL antibodies, the activity of PON1 was found to be dramatically decreased [19]. Charakida et al. [20] also confirmed the interactions between aCL antibodies and PON1. They showed that women with positive aPL antibodies have functional and structural arterial abnormalities that were associated with reduced activity of PON1. This implicates HDL and oxidative stress in the causal pathway for atherosclerosis in these patients. Moreover, in these patients, HDL has a “proatherogenic” phenotype by reducing nitric oxide bioavailability and impairing anti-inflammatory and antioxidant properties [20].
Finally, aCL seems to play an important role in promoting oxidative stress by inducing nitric oxide (NO) and superoxide production. This reaction favours enhanced production of plasma peroxynitrite, which is a powerful pro-oxidant substance. Indeed, in mice injected with aCL antibodies, there was an increase in serum nitrotyrosine suggesting that a permanent pro-oxidant environment induces the activation of iNOS and results in long-term downregulation of iNOS expression and subsequent endothelial dysfunction [21].
When oxidative stress is established, it contributes significantly to the pathophysiology of APS by (1) inducing protein structural modification, and (2) interfering with nitric oxide metabolism (Figure 2).
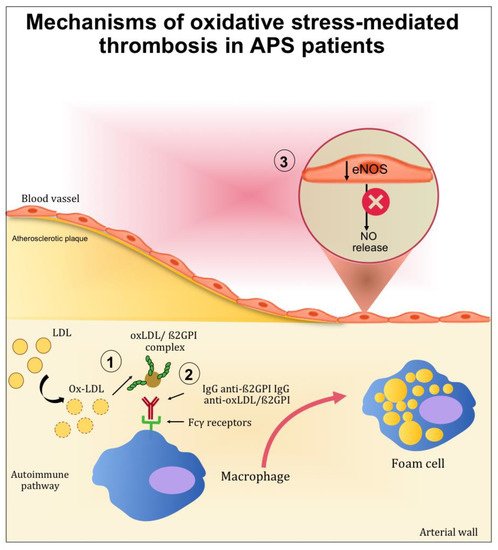
Figure 2. Mechanisms mediated by oxidative stress contributing to thrombotic complication in APS patients. (1) After oxidative modification, oxLDL binds β2GPI inside the arterial wall and further increases inflammation, oxidation, and cell activation. (2) Autoantibodies to this complex are produced resulting in circulating complexes (oxLDL/β2GPI/antibody). In the presence of anti-oxLDL/β2GPI antibodies, the uptake of oxLDL/β2GPI complexes by macrophage is increased and may further accelerate the development of atherosclerosis. (3) The endothelial nitric oxide synthase (eNOS) in the endothelial cells is inactivated, reducing the nitric oxide (NO) bioavailability.
2.1. The Role of Oxidative-Mediated Modifications
Clinical and epidemiological studies suggested that the presence of anti-β2GPI antibodies confers a significant risk of thrombosis, morbidity, and mortality in young adults [22]. For this reason, anti-β2GPI antibodies have been widely investigated to better understand the pathophysiology of APS and its complications.
β2GPI is a 50 kDa protein synthesized as a single polypeptide chain. It is mainly produced in the liver and may be detected in the blood at a concentration of 200 µg/mL. [23]. β2GPI has a role in coagulation, fibrinolysis, angiogenesis, and apoptosis [24]. Oxidation and nitrosylation of redox-sensitive cysteine residues are characteristic post-translational modifications of β2GPI occurring under conditions of increased oxidative or nitrosative stress. In particular, modifications of the sulfhydryl group (SH) alter the function of proteins containing cysteines in their catalytic domain or as interface residues of interacting proteins. ROS readily react with cysteine residues, especially redox-active cysteines, to form reversible or irreversible oxidized forms.
S-nitrosylation refers to a chemical reaction that occurs spontaneously or enzymatically in the presence of high NO concentrations. As a covalent post-translational modification on the cysteine thiol residue, s-nitrosylation has emerged as an important mechanism for functional regulation of most or all main classes of protein and intracellular processes [25]. These post-translational modifications directly affect the function of β2GPI and also confer an increase in the immunogenicity of β2GPI. In particular, the oxidation of β2GPI may increase the immunogenicity of the molecule by (1) increasing the affinity of anti-β2GPI antibodies to oxidised β2GPI; (2) causing immature monocyte-derived dendritic cells to mature that secret interleukin (IL)-12, IL-1, IL-6, IL-8, tumour necrosis factor-α and IL-10; (3) breaking immune tolerance [26].
The contribution of ROS to the development of APS has been studied in the context of lipid peroxidation and the formation of oxidized LDL (oxLDL)/β2GPI complexes. Indeed, patients with systemic autoimmune diseases displayed increased lipid peroxidation and oxLDL production [27][28]. After oxidative modification, electrostatic forces initially mediate the bind between oxLDL and β2GPI. After this initial interaction, more stable complexes (non-dissociable) are formed and stabilized by covalent interactions. These complexes are both proatherogenic and immunogenic. Indeed, the binding of β2GPI to oxLDL may occur inside the intima microenvironment of the arterial wall and further increase inflammation, oxidation, cell activation, and macrophage uptake of oxLDL/β2GPI complexes [29]. Moreover, patients with SLE and APS produce autoantibodies to this complex [30], and the resulting circulating immune complexes (oxLDL/β2GPI/antibody) may further accelerate the development of atherosclerosis. This was demonstrated in vitro by the increased uptake of oxLDL/β2GPI complexes by macrophage in the presence of anti-oxLDL/β2GPI antibodies [31][32]. These results provide an explanation for the accelerated development of atherosclerosis in autoimmune patients.
2.2. Role of Oxidative Stress in Nitric Oxide Metabolism
Among the mechanisms potentially implicated in oxidative stress-mediated atherothrombotic complications in APS, the inactivation of endothelial nitric oxide synthase (eNOS) is one of the most studied. eNOS is the predominant NOS isoform in the vasculature and is responsible for most of the nitric oxide (NO) produced in this tissue. NO is a short-lived gas molecule that is responsible for different biological actions in multiple tissues and cell types and is synthesized by eNOS to preserve vascular homeostasis [33]. Moreover, it exerts an atheroprotective function, and it inhibits blood clots and platelets adhesion to the endothelium [34].
A hypothetical connection between APS and alterations in the NO bioavailability has been evaluated in several studies performed both in mice and humans [35][36][37][38]. The evidence resulting from these studies showed a direct connection between the altered production of NO and APS pathogenesis.
In patients with aPL, a negative correlation was found between urinary NO metabolites (NOx) and IgG anticardiolipin, suggesting that aPL can negatively affect NO physiological activities [39].
In mice, the injections of polyclonal aPL and β2GPI monoclonal antibodies isolated from human patients can reduce the plasma concentration of NO metabolites. Moreover, the injection of aPL suppressed eNOS-mediated vascular relaxation by acetylcholine. Finally, in mice lacking eNOS, the increase in leukocyte adhesion to vascular endothelium and thrombus formation induced by aPL was not observed [36].
In vitro studies carried out by Ramesh et al. showed the impact of aPL in endothelial cells. In mice, aPL produced an increase of monocyte adhesion to endothelial cells, which is a mechanism directly related to atherosclerosis [36]. The same authors examined the role of β2GPI in aPL antagonism to eNOS by experiments that alternately included and excluded β2GPI from the surface of endothelial cells. When these cells were deprived of β2GPI, aPL did not cause eNOS inhibition, indicating that β2GPI is required for aPL full functioning [36][37][38].
3. Oxidative Stress in APS: Clinical and Experimental Studies
Several clinical studies evaluated oxidative stress biomarkers in APS patients (Table 1).
Table 1. Clinical, experimental, and in vitro studies describing changes of biomarkers of oxidative stress in APS patients.
| HUMAN STUDIES | |||
|---|---|---|---|
| Author/(Year)/[Reference] | Study Type (Setting) | Markers of Oxidative Stress | Main Results vs. Controls |
| Lambert et al., (2000) [19] | n = 56 APS patients n = 71 HS |
PON1 MDA-LDL |
↓ PON1 ↑ MDA-LDL |
| Delgado Alves et al., (2002) [40] | Cross-sectional study n = 32 SLE n = 36 with PAPS n = 20 controls |
HDL cholesterol PON activity TAC |
↓ HDL ↑ anti-HDL antibodies ↓ PON activity ↓ TAC |
| Ferro et al., (2003) [41] | n = 13 APL patients n = 11 negative APL patients |
Isoprostane | ↑ 8-isoprostane |
| Matsuura et al., (2006) [42] | n = 93 APS patients n = 161 HS |
oxLDL/beta2GPI | ↑ oxLDL/beta2GPI |
| Sciascia et al., (2012) [43] | n = 45 APS patients n = 75 HS |
Isoprostanes | ↑ 8-isoprostane ↑ Prostaglandin E2 (PGE) |
| Perez-Sanchez et al., (2015) [44] | n = 126 APS patients n = 61 HS |
TAC MnSOD Catalase GPx |
↓ TAC ↑ MnSOD ↑ Catalase ↓ GPx |
| Stanisavljevic et al., (2016) [45] | Cross-sectional case–control n = 140 APS patients n = 40 HS |
LOOH AOPP tSHG PON1 |
↔ LOOH ↑ AOPP ↓ tSHG ↓ PON1 |
| Lai et al., (2015) [46] | n = 12 APS patients n = 54 HS |
mitochondrial mass O2− production mTOR and FoxP3 expression |
↑ mitochondrial mass ↑ O2− production ↔ mTOR expression ↓ FoxP3 expression |
| Ibrahim (2017) [47] | n = 75 APS patients n = 120 HS |
polymorphisms of the PON1 | ↔ PON1 polymorphisms |
| Nojima et al., (2020) [48] | n = 58 APS patients n = 312 HS |
OSI | ↑ OSI |
| EXPERIMENTAL STUDIES | |||
| Delgado Alves et al., (2005) [49] | mice with SCID+ aCL and anti-aβ2-GPI monoclonal antibodies |
PON activity TAC |
↓ PON activity ↓ TAC |
| Benhamou et al., (2015) [50] | APS mice | gp91phox mRNA GSH/GSSH ratio |
↑ gp91phox mRNA ↑ left ventricular GSH/GSSH |
| Ding et al., (2015) [51] | APS mice wild-type mice |
p47phox | ↑ p47phox mRNA ↑ p47phox phosphorylation |
| IN VITRO STUDIES | |||
| Ferro et al., (2003) [41] | Human monocytes from (HS) and anti-β2GP1 antibodies (50, 100, 200 µg/mL) |
O2− production | ↑ O2− production |
| Simoncini et al., (2005) [52] | HUVEC and IgG (IgG-APS) from 12 APS patients |
ROS production | ↑ ROS production MAP kinases pathway: ↑ p38 phosphorylation ↑ ATF-2 |
Abbreviations: healthy subjects (HS); Human umbilical vein endothelial cells (HUVEC); Mitogen-activated protein (MAP) kinases; activating transcription factor-2 (ATF-2); Oxidized low-density lipoprotein (oxLDL); b2-glycoprotein I (2GPI); prostaglandin E2 (PGE); Manganese-SOD, total antioxidant capacity (TAC); manganese-superoxide dismutase (MnSOD); glutathione peroxidase (GPx); lipid hydroperoxydes (LOOH); advanced oxidation protein products (AOPP); total sulfhydryl groups (tSHG); paraoxonase 1 activity (PON1); oxidation stress index (OSI); severe combined immunodeficiency (SCID); anticardiolipin (aCL); Total antioxidant capacity (TAC); ↑ increase; ↓ decrease; ↔ no changes.
aPLs have been demonstrated to induce an increased expression of molecules able to produce an expanded oxidative status in plasma, as demonstrated by high levels of prostaglandin F2-isoprostanes in APS patients. F2-isoprostanes are arachidonic acid products formed on membrane phospholipids by the action of ROS. As F2-isoprostanes are characterized by stability and specificity for lipid peroxidation, they represent a reliable marker for quantitative measurement of lipid peroxidation oxidative stress in vivo and prediction of cardiovascular events [53][54]. Specifically, a study conducted on 45 APS patients found higher values of 8-isoprostanes in the APS group than in the other groups. Moreover, APS patients with enhanced inflammation and oxidative stress recorded more thrombotic events compared to the control group (69% vs. 6.5%). Similar results were shown by another study that investigates the relationship between oxidative stress and monocyte tissue factor (TF) expression in a cross-sectional comparison of aPL-positive and aPL-negative patients [41]. In fact, in these patients, an upregulation of monocyte TF expression was associated with thrombosis [55]. The results showed that compared with aPL-negative subjects, in aPL-positive patients higher values of isoprostanes and monocyte TF antigen and activity were observed [41].
PON1 is a hydrolytic enzyme with a wide range of substrates, and the capability to protect against lipid oxidation. There is considerable in vitro and in vivo data that prove the beneficial effects of PON1 in several atherosclerosis-related processes [56].
In an observational study, among 56 patients with APS, 37 presented arterial thrombosis, 16 presented venous thrombosis and all showed malondialdehyde-modified LDL (MDA-LDL) at significantly higher levels than controls. Furthermore, basal serum of PON1 activity was dramatically decreased in a subgroup of patients in comparison with the controls [19]. These results suggest that PON1 abnormalities that play a role in the APS might be associated with a higher risk of arterial thrombosis. Genetic analysis confirmed the role of PON1 in APS. In fact, PON1 L55M polymorphism resulted in an association with APS [47].
Consistently with these results, a cross-sectional study showed that PON1 is reduced in 36 patients with primary APS compared with 20 healthy subjects (HS) [40]. Additionally, the total antioxidant capacity (TAC), which quantifies the overall antioxidant defence of plasma, analysed in patients with primary APS did not differ significantly from levels in the control group but correlated positively with PON activity [40].
Chronic, autoimmune, vascular inflammation together with decreased PON activity may contribute to oxidative stress, LDL modification (oxLDL) and oxLDL/2GPI complex formation that reflect the oxidative stress degree. Matsuura et al. revealed that serum levels of IgG anti-oxLDL/2GPI antibodies were significantly higher in systemic lupus erythematosus (SLE) patients with APS compared to SLE controls without APS. In addition, high concentrations of these IgG antibodies were observed in APS patients with a history of arterial thrombosis. Therefore, the presence of circulating oxLDL/2GPI complexes and IgG antibodies to these complexes indicates significant vascular damage and oxidative stress as well as a significant role in autoimmune-mediated atherothrombosis [42].
A cross-sectional case–control clinical study, including a total of 180 patients with primary and secondary APS and a control group, investigated several oxidative stress markers of endothelial damage measured by flow-mediated dilation (FMD). Biomarkers of oxidative stress, lipid hydroperoxydes (LOOH), advanced oxidation protein products (AOPP), total sulfhydryl groups (tSHG), and PON1 activity resulted altered in APS patients [45].
In addition to the evidence from human models, some studies on murine models supported that enhanced oxidative stress occurs in APS and the role of oxidant/antioxidant balance.
Severe combined immunodeficiency (SCID) mice were injected with Hybridomas producing human and murine aCL antibodies and β2GPI monoclonal antibodies. Results showed that PON1 activity, NO levels, and expression of total antioxidant capacity (TAC) were reduced. Conversely, peroxynitrite and superoxide concentrations in plasma were increased. These data confirm that aCL antibodies are associated with the decreased PON activity and reduced endothelial function that may occur in the APS [49].
At the molecular level, it has been demonstrated that in the liver of the APS mouse model, both mRNA and protein expression of 47phox, a protein involved in the upregulation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, were increased compared with the control group [51]. As NADPH oxidase is the main source of ROS [57], the results suggest that NADPH oxidase-mediated oxidative stress leads to endothelial cell injury in APS.
The studies described so far analyse the oxidative stress at plasmatic levels. Perez-Sanchez et al. studied oxidative stress at the cellular level by analysing biomarkers in circulating leucocytes from APS patients. Higher peroxide production, the nuclear abundance of Nrf2, antioxidant enzymatic activity, decreased intracellular glutathione, and altered mitochondrial membrane potential were found in monocytes and neutrophils from APS patients compared to healthy subjects [58]. Specifically, ROS production was markedly increased in monocytes and neutrophils of APS patients compared with healthy donors, as was the expression of Nrf2, the main regulator of antioxidant genes. Moreover, intracellular reduced GSH was significantly decreased in both cell types and the activities of catalase (CAT) and glutathione peroxidase (GPx) resulted in being strongly reduced. Furthermore, a significant systemic reduction of total antioxidant capacity (TAC) in plasma from APS patients was found compared to healthy donors and might indicate a reduced ability to counteract ROS production and oxidative damage [58].
According to the results of these studies, showing that oxidative stress is directly involved in the pathophysiology of atherothrombosis in APS, the evaluation of oxidative stress biomarkers could be used as serologic indicators to assess the APS patient’s risk for vascular complications. Moreover, vascular, preventive strategies and more targeted therapeutic interventions should be developed.
References
- Ruiz-Irastorza, G.; Crowther, M.; Branch, W.; Khamashta, M.A. Antiphospholipid syndrome. Lancet 2010, 376, 1498–1509.
- Willis, R.; Pierangeli, S.S. Pathophysiology of the antiphospholipid antibody syndrome. Autoimmun. Highlights 2011, 2, 35–52.
- Pastori, D.; Ames, P.R.J.; Triggiani, M.; Ciampa, A.; Cammisotto, V.; Carnevale, R.; Pignatelli, P.; Bucci, T. Antiphospholipid antibodies and heart failure with preserved ejection fraction. The multicenter athero-aps study. J. Clin. Med. 2021, 10, 3180.
- Abreu, M.M.; Danowski, A.; Wahl, D.G.; Amigo, M.C.; Tektonidou, M.; Pacheco, M.S.; Fleming, N.; Domingues, V.; Sciascia, S.; Lyra, J.O.; et al. The relevance of “non-criteria” clinical manifestations of antiphospholipid syndrome: 14th International Congress on Antiphospholipid Antibodies Technical Task Force Report on Antiphospholipid Syndrome Clinical Features. Autoimmun. Rev. 2015, 14, 401–414.
- Bucci, T.; Menichelli, D.; Pignatelli, P.; Triggiani, M.; Violi, F.; Pastori, D. Relationship of Antiphospholipid Antibodies to Risk of Dementia: A Systematic Review. J. Alzheimer’s Dis. 2019, 69, 561–576.
- Meroni, P.L.; Borghi, M.O.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339.
- De Jesús, G.R.; Benson, A.E.; Chighizola, C.B.; Sciascia, S.; Branch, D.W. 16th International Congress on Antiphospholipid Antibodies Task Force Report on Obstetric Antiphospholipid Syndrome. Lupus 2020, 29, 1601–1615.
- Pignatelli, P.; Ettorre, E.; Menichelli, D.; Pani, A.; Violi, F.; Pastori, D. Seronegative antiphospholipid syndrome: Refining the value of “non-criteria” antibodies for diagnosis and clinical management. Haematologica 2020, 105, 562–572.
- El Hasbani, G.; Taher, A.T.; Sciascia, S.; Uthman, I. Antiphospholipid syndrome: The need for new international classification criteria. Expert Rev. Clin. Immunol. 2021, 17, 385–394.
- Pastori, D.; Misasi, R.; Sorice, M.; Cribari, F.; Menichelli, D.; Violi, F.; Pignatelli, P. Multiple Arterial Thrombosis in Seronegative Antiphospholipid Syndrome: Need for New Diagnostic Criteria? Eur. J. Case Reports Intern. Med. 2019, 6, 1.
- Cervera, R.; Serrano, R.; Pons-Estel, G.J.; Ceberio-Hualde, L.; Shoenfeld, Y.; De Ramón, E.; Buonaiuto, V.; Jacobsen, S.; Zeher, M.M.; Tarr, T.; et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: A multicentre prospective study of 1000 patients. Ann. Rheum. Dis. 2015, 74, 1011–1018.
- Carnevale, R.; Pignatelli, P.; Lenti, L.; Buchetti, B.; Sanguigni, V.; Di Santo, S.; Violi, F. LDL are oxidatively modified by platelets via GP91(phox) and accumulate in human monocytes. FASEB J. 2007, 21, 927–934.
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116.
- Violi, F.; Loffredo, L.; Carnevale, R.; Pignatelli, P.; Pastori, D. Atherothrombosis and Oxidative Stress: Mechanisms and Management in Elderly. Antioxid. Redox Signal. 2017, 27, 1083–1124.
- Conti, V.; Forte, M.; Corbi, G.; Russomanno, G.; Formisano, L.; Landolfi, A.; Izzo, V.; Filippelli, A.; Vecchione, C.; Carrizzo, A. Sirtuins: Possible Clinical Implications in Cardio and Cerebrovascular Diseases. Curr. Drug Targets 2016, 18, 473–484.
- Miceli, M.; Roma, E.; Rosa, P.; Feroci, M.; Loreto, M.A.; Tofani, D.; Gasperi, T. Synthesis of Benzofuran-2-one derivatives and evaluation of their antioxidant capacity by comparing DPPH assay and cyclic voltammetry. Molecules 2018, 23, 710.
- Nocella, C.; Cammisotto, V.; Pigozzi, F.; Borrione, P.; Fossati, C.; D’amico, A.; Cangemi, R.; Peruzzi, M.; Gobbi, G.; Ettorre, E.; et al. Impairment between Oxidant and Antioxidant Systems: Short- and Long-term Implications for Athletes’ Health. Nutrients 2019, 11, 1353.
- Gergely, P.; Grossman, C.; Niland, B.; Puskas, F.; Neupane, H.; Allam, F.; Banki, K.; Phillips, P.E.; Perl, A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 175–190.
- Lambert, M.; Boullier, A.; Hachulla, E.; Fruchart, J.C.; Teissier, E.; Hatron, P.Y.; Duriez, P. Paraoxonase activity is dramatically decreased in patients positive for anticardiolipin antibodies. Lupus 2000, 9, 299–300.
- Charakida, M.; Besler, C.; Batuca, J.R.; Sangle, S.; Marques, S.; Sousa, M.; Wang, G.; Tousoulis, D.; Delgado Alves, J.; Loukogeorgakis, S.P.; et al. Vascular abnormalities, paraoxonase activity, and dysfunctional HDL in primary antiphospholipid syndrome. JAMA—J. Am. Med. Assoc. 2009, 302, 1210–1217.
- Alves, J.D.; Grima, B. Oxidative stress in systemic lupus erythematosus and antiphospholipid syndrome: A gateway to atherosclerosis. Curr. Rheumatol. Rep. 2003, 5, 383–390.
- Sciascia, S.; Baldovino, S.; Schreiber, K.; Solfietti, L.; Radin, M.; Cuadrado, M.J.; Menegatti, E.; Erkan, D.; Roccatello, D. Thrombotic risk assessment in antiphospholipid syndrome: The role of new antibody specificities and thrombin generation assay. Clin. Mol. Allergy 2016, 14, 6.
- Lozier, J.; Takahashi, N.; Putnam, F.W. Complete amino acid sequence of human plasma β2-glycoprotein I. Proc. Natl. Acad. Sci. USA 1984, 81, 3640–3644.
- Nakagawa, H.; Yasuda, S.; Miyazaki, T. Novel Function of Beta 2 Glycoprotein I in Angiogenesis. Curr. Angiogenes. 2015, 3, 132–138.
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166.
- Passam, F.H.; Giannakopoulos, B.; Mirarabshahi, P.; Krilis, S.A. Molecular pathophysiology of the antiphospholipid syndrome: The role of oxidative post-translational modification of beta 2 glycoprotein I. J. Thromb. Haemost. 2011, 9, 275–282.
- Morgan, P.E.; Sturgess, A.D.; Davies, M.J. Increased levels of serum protein oxidation and correlation with disease activity in systemic lupus erythematosus. Arthritis Rheum. 2005, 52, 2069–2079.
- Frostegård, J.; Svenungsson, E.; Wu, R.; Gunnarsson, I.; Lundberg, I.E.; Klareskog, L.; Hörkkö, S.; Witztum, J.L. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005, 52, 192–200.
- Matsuura, E.; Lopez, L. Autoimmune-mediated atherothrombosis. Lupus 2008, 17, 879–888.
- Kobayashi, K.; Kishi, M.; Atsumi, T.; Bertolaccini, M.L.; Makino, H.; Sakairi, N.; Yamamoto, I.; Yasuda, T.; Khamashta, M.A.; Hughes, G.R.V.; et al. Circulating oxidized LDL forms complexes with β 2-glycoprotein I: Implication as an atherogenic autoantigen. J. Lipid Res. 2003, 44, 716–726.
- Hasunuma, Y.; Matsuura, E.; Makita, Z.; Katahira, T.; Nishi, S.; Koike, T. Involvement of beta2-glycoprotein I and anticardiolipin antibodies in oxidatively modified low-density lipoprotein uptake by macrophages. Clin. Exp. Immunol. 1997, 107, 569–573.
- Kobayashi, K.; Matsuura, E.; Liu, Q.; Furukawa, J.I.; Kaihara, K.; Inagaki, J.; Atsumi, T.; Sakairi, N.; Yasuda, T.; Voelker, D.R.; et al. A specific ligand for β2-glycoprotein I mediates autoantibody-dependent uptake of oxidized low density lipoprotein by macrophages. J. Lipid Res. 2001, 42, 697–709.
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Loscalzo, J. Jin Vascular nitric oxide: Formation and function. J. Blood Med. 2010, 1, 147.
- Ames, P.R.J.; Tommasino, C.; Alves, J.; Morrow, J.D.; Iannaccone, L.; Fossati, G.; Caruso, S.; Caccavo, F.; Brancaccio, V. Antioxidant susceptibility of pathogenic pathways in subjects with antiphospholipid antibodies: A pilot study. Lupus 2000, 9, 688–695.
- Ramesh, S.; Morrell, C.N.; Tarango, C.; Thomas, G.D.; Yuhanna, I.S.; Girardi, G.; Herz, J.; Urbanus, R.T.; De Groot, P.G.; Thorpe, P.E.; et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via β2GPI and apoER2. J. Clin. Investig. 2011, 121, 120–131.
- Urbanus, R.T.; Pennings, M.T.T.; Derksen, R.H.W.M.; de Groot, P.G. Platelet activation by dimeric β2-glycoprotein I requires signaling via both glycoprotein Ibα and apolipoprotein E receptor 2′. J. Thromb. Haemost. 2008, 6, 1405–1412.
- Lutters, B.C.H.; Derksen, R.H.W.M.; Tekelenburg, W.L.; Lenting, P.J.; Arnout, J.; De Groot, P.G. Dimers of β2-Glycoprotein I Increase Platelet Deposition to Collagen via Interaction with Phospholipids and the Apolipoprotein E Receptor 2′. J. Biol. Chem. 2003, 278, 33831–33838.
- Ames, P.R.J.; Batuca, J.R.; Ciampa, A.; Iannaccone, L.; Alves, J.D. Clinical relevance of nitric oxide metabolites and nitrative stress in thrombotic primary antiphospholipid syndrome. J. Rheumatol. 2010, 37, 2523–2530.
- Ramesh, S.; Morrell, C.N.; Tarango, C.; Thomas, G.D.; Yuhanna, I.S.; Girardi, G.; Herz, J.; Urbanus, R.T.; De Groot, P.G.; Thorpe, P.E.; et al. Antibodies to high-density lipoprotein and beta2-glycoprotein I are inversely correlated with paraoxonase activity in systemic lupus erythematosus and primary antiphospholipid syndrome. Arthritis Rheum. 2002, 46, 2686–2694.
- Ferro, D.; Saliola, M.; Meroni, P.L.; Valesini, G.; Caroselli, C.; Praticó, D.; Fitzgerald, G.A.; Shoenfeld, Y.; Violi, F. Enhanced monocyte expression of tissue factor by oxidative stress in patients with antiphospholipid antibodies: Effect of antioxidant treatment. J. Thromb. Haemost. 2003, 1, 523–531.
- Matsuura, E.; Kobayashi, K.; Hurley, B.L.; Lopez, L.R. Atherogenic oxidized low-density lipoprotein/β2- glycoprotein I (oxLDL/β2GPI) complexes in patients with systemic lupus erythematosus and antiphospholipid syndrome. Lupus 2006, 15, 478–483.
- Sciascia, S.; Roccatello, D.; Bertero, M.T.; Di Simone, D.; Cosseddu, D.; Vaccarino, A.; Bazzan, M.; Rossi, D.; Garcia-Fernandez, C.; Ceberio, L.; et al. 8-Isoprostane, prostaglandin E2, C-reactive protein and serum amyloid A as markers of inflammation and oxidative stress in antiphospholipid syndrome: A pilot study. Inflamm. Res. 2012, 61, 809–816.
- Perez-Sanchez, C.; Barbarroja, N.; Messineo, S.; Ruiz-Limon, P.; Rodriguez-Ariza, A.; Jimenez-Gomez, Y.; Khamashta, M.A.; Collantes-Estevez, E.; Cuadrado, M.J.; Aguirre, M.A.; et al. Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann. Rheum. Dis. 2015, 74, 1441–1449.
- Stanisavljevic, N.; Stojanovich, L.; Marisavljevic, D.; Djokovic, A.; Dopsaj, V.; Kotur-Stevuljevic, J.; Martinovic, J.; Memon, L.; Radovanovic, S.; Todic, B.; et al. Lipid peroxidation as risk factor for endothelial dysfunction in antiphospholipid syndrome patients. Clin. Rheumatol. 2016, 35, 2485–2493.
- Lai, Z.-W.; Marchena-Mendez, I.; Perl, A. Oxidative stress and Treg depletion in lupus patients with anti-phospholipid syndrome. Clin. Immunol. 2015, 158, 148–152.
- Ibrahim, A.A.; El-Lebedy, D.; Ashmawy, I.; Hady, M.A. Association between paraoxonase-1 gene Q192R and L55M polymorphisms in systemic lupus erythematosus (SLE) and anti-phospholipid syndrome (APS) in a population from Cairo of Egypt. Clin. Rheumatol. 2017, 36, 1305–1310.
- Nojima, J.; Kaneshige, R.; Motoki, Y.; Ieko, M. Increased oxidative stress may be a risk factor for thromboembolic complications in patients with antiphospholipid syndrome. Thromb. Res. 2020, 196, 52–53.
- Delgado Alves, J.; Mason, L.J.; Ames, P.R.J.; Chen, P.P.; Rauch, J.; Levine, J.S.; Subang, R.; Isenberg, D.A. Antiphospholipid antibodies are associated with enhanced oxidative stress, decreased plasma nitric oxide and paraoxonase activity in an experimental mouse model. Rheumatology 2005, 44, 1238–1244.
- Benhamou, Y.; Miranda, S.; Armengol, G.; Harouki, N.; Drouot, L.; Zahr, N.; Thuillez, C.; Boyer, O.; Levesque, H.; Joannides, R.; et al. Infliximab improves endothelial dysfunction in a mouse model of antiphospholipid syndrome: Role of reduced oxidative stress. Vascul. Pharmacol. 2015, 71, 93–101.
- Ding, X.; Yang, Z.; Han, Y.; Yu, H. Correlation of long-chain fatty acid oxidation with oxidative stress and inflammation in pre-eclampsia-like mouse models. Placenta 2015, 36, 1442–1449.
- Simoncini, S. Role of reactive oxygen species and p38 MAPK in the induction of the pro-adhesive endothelial state mediated by IgG from patients with anti-phospholipid syndrome. Int. Immunol. 2005, 17, 489–500.
- Milne, G.L.; Yin, H.; Hardy, K.D.; Davies, S.S.; Roberts, L.J. Isoprostane generation and function. Chem. Rev. 2011, 111, 5973–5996.
- Pignatelli, P.; Pastori, D.; Carnevale, R.; Farcomeni, A.; Cangemi, R.; Nocella, C.; Bartimoccia, S.; Vicario, T.; Saliola, M.; Lip, G.Y.H.; et al. Serum NOX2 and urinary isoprostanes predict vascular events in patients with atrial fibrillation. Thromb. Haemost. 2015, 113, 617–624.
- Amengual, O.; Atsumi, T.; Khamashta, M.A.; Hughes, G.R. The role of the tissue factor pathway in the hypercoagulable state in patients with the antiphospholipid syndrome. Thromb. Haemost. 1998, 79, 276–281.
- Litvinov, D.; Mahini, H.; Garelnabi, M. Antioxidant and anti-inflammatory role of paraoxonase 1: Implication in arteriosclerosis diseases. N. Am. J. Med. Sci. 2012, 4, 523–532.
- Manea, A. NADPH oxidase-derived reactive oxygen species: Involvement in vascular physiology and pathology. Cell Tissue Res. 2010, 342, 325–339.
- Perez-Sanchez, C.; Ruiz-Limon, P.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodriguez-Ariza, A.; Segui, P.; Collantes-Estevez, E.; Barbarroja, N.; Khraiwesh, H.; et al. Mitochondrial dysfunction in antiphospholipid syndrome: Implications in the pathogenesis of the disease and effects of coenzyme Q 10 treatment. Blood 2012, 119, 5859–5870.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
3 times
(View History)
Update Date:
07 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No