Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Neeraj Kumar | + 3518 word(s) | 3518 | 2021-11-18 07:02:44 | | | |
| 2 | Camila Xu | -1915 word(s) | 1603 | 2021-12-07 07:53:11 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kumar, N. Autonomous Molecular Design: Machine Intelligence. Encyclopedia. Available online: https://encyclopedia.pub/entry/16801 (accessed on 18 July 2026).
Kumar N. Autonomous Molecular Design: Machine Intelligence. Encyclopedia. Available at: https://encyclopedia.pub/entry/16801. Accessed July 18, 2026.
Kumar, Neeraj. "Autonomous Molecular Design: Machine Intelligence" Encyclopedia, https://encyclopedia.pub/entry/16801 (accessed July 18, 2026).
Kumar, N. (2021, December 06). Autonomous Molecular Design: Machine Intelligence. In Encyclopedia. https://encyclopedia.pub/entry/16801
Kumar, Neeraj. "Autonomous Molecular Design: Machine Intelligence." Encyclopedia. Web. 06 December, 2021.
Copy Citation
The workflow for computational autonomous molecular design (CAMD) must be an integrated and closed-loop system with (i) efficient data generation and extraction tools, (ii) robust data representation techniques, (iii) physics-informed predictive machine learning (ML) models, and (iv) tools to generate new molecules using the knowledge learned from steps i–iii.
autonomous workflow
therapeutic design
computer aided drug discovery
computational modeling and simulations
1. Components of Computational Autonomous Molecular Design Workflow
The workflow for computational autonomous molecular design (CAMD) must be an integrated and closed-loop system (Figure 1) with: (i) efficient data generation and extraction tools, (ii) robust data representation techniques, (iii) physics-informed predictive machine learning models, and (iv) tools to generate new molecules using the knowledge learned from steps i–iii. Ideally, an autonomous computational workflow for molecule discovery would learn from its own experience and adjust its functionality as the chemical environment or the targeted functionality changes through active learning. This can be achieved when all the components work in collaboration with each other, providing feedback while improving model performance as we move from one step to another.
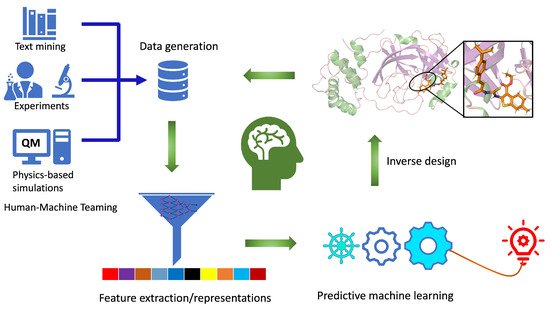
Figure 1. Closed-loop workflow for computational autonomous molecular design (CAMD) for medical therapeutics. Individual components of the workflow are labeled. It consists of data generation, feature extraction, predictive machine learning, and an inverse molecular design engine.
For data generation in CAMD, high-throughput density functional theory (DFT) [1][2] is a common choice mainly because of its reasonable accuracy and efficiency [3][4]. In DFT, we typically feed in 3D structures to predict the properties of interest. Data generated from DFT simulations are processed to extract the more relevant structural and properties data, which are then either used as input to learning the representation [5][6] or as a target required for the ML models [7][8][9]. Data generated can be used in two different ways: to predict the properties of new molecules using a direct supervised ML approach and to generate new molecules with the desired properties of interest using inverse design. CAMD can be tied with supplementary components, such as databases, to store the data and visualize it. The AI-assisted CAMD workflow presented here is the first step in developing automated workflows for molecular design. Such an automated pipeline will not only accelerate the hit identification and lead optimization for the desired therapeutic candidates but can actively be used for machine reasoning to develop transparent and interpretable ML models. These workflows, in principle, can be combined intelligently with experimental setups for computer-aided synthesis or screening planning that includes synthesis and characterization tools, which are expensive to explore in the desired chemical space. Instead, experimental measurements and characterization should be performed intelligently for only the AI-designed lead compounds obtained from CAMD.
The data generated from inverse design in principle should be validated by using an integrated DFT method for the desired properties or by high throughput docking with a target protein to find out its affinity in the closed-loop system, then accordingly update the rest of the CAMD. These steps are then repeated in a closed-loop, thus improving and optimizing the data representation, property prediction, and new data generation component. Once we have confidence in our workflow to generate valid new molecules, the validation step with DFT can be bypassed or replaced with an ML predictive tool to make the workflow computationally more efficient. In the following, we briefly discuss the main component of the CAMD, while reviewing the recent breakthroughs achieved.
2. Data Generation and Molecular Representation
ML models are data-centric—the more data, the better the model performance. A lack of accurate, ethically sourced well-curated data is the major bottleneck limiting their use in many domains of physical and biological science. For some sub-domains, a limited amount of data exists that comes mainly from physics-based simulations in databases [10][11] or from experimental databases, such as NIST [12]. For other fields, such as for biochemical reactions [13], we have databases with the free energy of reactions, but they are obtained with empirical methods, which are not considered ideal as ground truth for machine learning models. For many domains, accurate and curated data does not exist. In these scenarios, slightly unconventional yet very effective approaches to creating data from published scientific literature and patents for ML have recently gained adoption [14][15][16][17]. These approaches are based on natural language processing (NLP) to extract chemistry and biology data from open sources published literature. Developing a cutting-edge NLP-based tool to extract, learn, and the reason the extracted data would definitely reduce the timeline for high throughput experimental design in the lab. This would significantly expedite the decision-making based on the existing literature to set up future experiments in a semi-automated way. The resulting tools based on human-machine teaming are much needed for scientific discovery.
3. Molecular Representation in Automated Pipelines
A robust representation of molecules is required for the accurate functioning of the ML models [18]. An ideal molecular representation should be unique, invariant with respect to different symmetry operations, invertible, efficient to obtain and capture the physics, stereochemistry, and structural motif. Some of these can be achieved by using the physical, chemical, and structural properties [19], which, all together, are rarely well documented so obtaining this information is considered a cumbersome task. Over time, this has been tackled by using several alternative approaches that work well for specific problems [20][21][22][23][24][25] as shown in Figure 2. However, developing universal representations of molecules for diverse ML problems is still a challenging task, and any gold standard method that works consistently for all kinds of problems is yet to be discovered. Molecular representations primarily used in the literature falls into two broad categories: (a) 1D and/or 2D representations designed by experts using domain-specific knowledge, including properties from the simulation and experiments, and (b) iteratively learned molecular representations directly from the 3D nuclear coordinates/properties within ML frameworks.
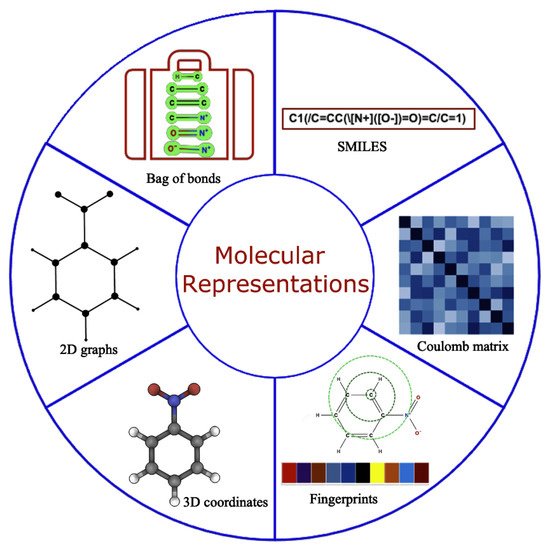
Figure 2. Molecular representation with all possible formulations used in the literature for predictive and generative modeling.
Expert-engineered molecular representations have been extensively used for predictive modeling in the last decade, which includes properties of the molecules [26][27], structured text sequences [28][29][30] (SMILES, InChI), molecular fingerprints [31], among others. Such representations are carefully selected for each specific problem using domain expertise, a lot of resources, and time. The SMILES representation of molecules is the main workhorse as a starting point for both representation learning as well as for generating expert-engineered molecular descriptors. For the latter, SMILES strings can be used directly as a one-hot encoded vector to calculate fingerprints or to calculate the range of empirical properties using different open-source platforms, such as RDkit [32] or ChemAxon [33], thereby bypassing expensive features generation from quantum chemistry/experiments by providing a faster speed and diverse properties, including 3D coordinates, for molecular representations. Moreover, SMILES can be easily converted into 2D graphs, which is the preferred choice to date for generative modeling, where molecules are treated as graphs with nodes and edges. Although significant progress has been made in molecular generative modeling using mainly SMILES strings [28], they often lead to the generation of syntactically invalid molecules and are synthetically unexplored. In addition, SMILES are also known to violate fundamental physics and chemistry-based constraints [34][35]. Case-specific solutions to circumvent some of these problems exist, but a universal solution is still unknown. The extension of SMILES was attempted by more robustly encoding rings and branches of molecules to find more concrete representations with high semantical and syntactical validity using canonical SMILES [36][37], InChI [29][30], SMARTS [38], DeepSMILES [39], DESMILES [40], etc. More recently, Kren et al. proposed a 100% syntactically correct and robust string-based representation of molecules known as SELFIES [34], which has been increasingly adopted for predictive and generative modeling [41].
Recently, molecular representations that can be iteratively learned directly from molecules have been increasingly adopted, mainly for predictive molecular modeling, achieving chemical accuracy for a range of properties [19][42][43]. Such representations as shown in Figure 3 are more robust and outperform expert-designed representations in drug design and discovery [44]. For representation learning, different variants of graph neural networks are a popular choice [22][45]. It starts with generating the atom (node) and bond (edge) features for all the atoms and bonds within a molecule, which are iteratively updated using graph traversal algorithms, taking into account the chemical environment information to learn a robust molecular representation. The starting atom and bond features of the molecule may just be a one-hot encoded vector to only include atom-type, bond-type, or a list of properties of the atom and bonds derived from SMILES strings. Yang et al. achieved the chemical accuracy for predicting a number of properties with their ML models by combining the atom and bond features of molecules with global state features before being updated during the iterative process [46].
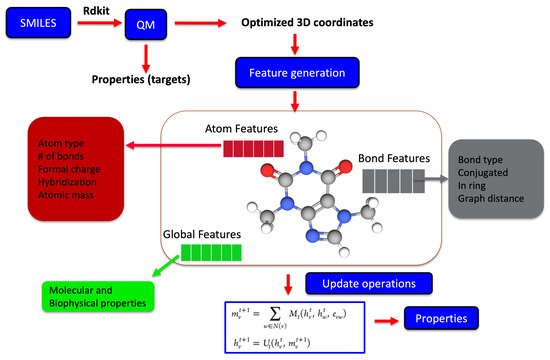
Figure 3. The iterative update process is used for learning a robust molecular representation either based on 2D SMILES or 3D optimized geometrical coordinates from physics-based simulations. The molecular graph is usually represented by features at the atomic level, bond level, and global state, which represent the key properties. Each of these features is iteratively updated during the representation learning phase, which is subsequently used for the predictive part of the model.
Molecules are 3D multiconformational entities, and hence, it is natural to assume that they can be well represented by the nuclear coordinates as is the case of physics-based molecular simulations [47]. However, with coordinates, the representation of molecules is non-invariant, non-invertible, and non-unique in nature [20] and hence not commonly used in conventional machine learning. In addition, the coordinates by themselves do not carry information about the key attribute of molecules, such as bond types, symmetry, spin states, charge, etc., in a molecule. Approaches/architectures have been proposed to create robust, unique, and invariant representations from nuclear coordinates using atom-centered Gaussian functions, tensor field networks, and, more robustly, by using representation learning techniques [19][43][48][49][50][51], as shown in Figure 3.
References
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871.
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138.
- Jain, A.; Hautier, G.; Moore, C.J.; Ong, S.P.; Fischer, C.C.; Mueller, T.; Persson, K.A.; Ceder, G. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 2011, 50, 2295–2310.
- Qu, X.; Jain, A.; Rajput, N.N.; Cheng, L.; Zhang, Y.; Ong, S.P.; Brafman, M.; Maginn, E.; Curtiss, L.A.; Persson, K.A. The Electrolyte Genome project: A big data approach in battery materials discovery. Comput. Mater. Sci. 2015, 103, 56–67.
- Qiao, Z.; Welborn, M.; Anandkumar, A.; Manby, F.R.; Miller, T.F. OrbNet: Deep learning for quantum chemistry using symmetry-adapted atomic-orbital features. J. Chem. Phys. 2020, 153, 124111.
- Lee, S.J.R.; Husch, T.; Ding, F.; Miller, T.F. Analytical Gradients for Molecular-Orbital-Based Machine Learning. arXiv 2020, arXiv:2012.08899.
- Dral, P.O. Quantum Chemistry in the Age of Machine Learning. J. Phys. Chem. Lett. 2020, 11, 2336–2347.
- Bogojeski, M.; Vogt-Maranto, L.; Tuckerman, M.E.; Müller, K.R.; Burke, K. Quantum chemical accuracy from density functional approximations via machine learning. Nat. Commun. 2020, 11, 5223.
- Joshi, R.P.; McNaughton, A.; Thomas, D.G.; Henry, C.S.; Canon, S.R.; McCue, L.A.; Kumar, N. Quantum Mechanical Methods Predict Accurate Thermodynamics of Biochemical Reactions. ACS Omega 2021, 6, 9948–9959.
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; von Lilienfeld, O.A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 2014, 1, 140022.
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875.
- Shen, V.; Siderius, D.; Krekelberg, W.; Mountain, R.D.; Hatch, H.W. NIST Standard Reference Simulation Website, NIST Standard Reference Database Number 173; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017.
- Seaver, S.M.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the Integration of Metabolic Annotations and the Reconstruction, Comparison and Analysis of Metabolic Models for Plants, Fungi and Microbes. Nucleic Acids Res. 2021, 49, D575–D588.
- Kononova, O.; Huo, H.; He, T.; Rong, Z.; Botari, T.; Sun, W.; Tshitoyan, V.; Ceder, G. Text-mined dataset of inorganic materials synthesis recipes. Sci. Data 2019, 6, 1–11.
- Zheng, S.; Dharssi, S.; Wu, M.; Li, J.; Lu, Z. Text Mining for Drug Discovery. In Bioinformatics and Drug Discovery; Larson, R.S., Oprea, T.I., Eds.; Springer: New York, NY, USA, 2019; pp. 231–252.
- Singhal, A.; Simmons, M.; Lu, Z. Text mining for precision medicine: Automating disease-mutation relationship extraction from biomedical literature. J. Am. Med. Inform. Assoc. 2016, 23, 766–772.
- Krallinger, M.; Rabal, O.; Lourenço, A.; Oyarzabal, J.; Valencia, A. Information Retrieval and Text Mining Technologies for Chemistry. Chem. Rev. 2017, 117, 7673–7761.
- Huang, B.; von Lilienfeld, O.A. Communication: Understanding molecular representations in machine learning: The role of uniqueness and target similarity. J. Chem. Phys. 2016, 145, 161102.
- Chen, C.; Ye, W.; Zuo, Y.; Zheng, C.; Ong, S.P. Graph Networks as a Universal Machine Learning Framework for Molecules and Crystals. Chem. Mater. 2019, 31, 3564–3572.
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849.
- Bjerrum, E.J. SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arXiv 2017, arXiv:1703.07076.
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural Message Passing for Quantum Chemistry. arXiv 2017, arXiv:1704.01212.
- Hamilton, W.L.; Ying, R.; Leskovec, J. Representation Learning on Graphs: Methods and Applications. arXiv 2017, arXiv:1709.05584.
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. Aided Mol. Des. 2016, 30, 595–608.
- Wu, Z.; Ramsundar, B.; Feinberg, E.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530.
- Rupp, M.; Tkatchenko, A.; Muller, K.R.; Von Lilienfeld, O.A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108, 058301.
- Hansen, K.; Biegler, F.; Ramakrishnan, R.; Pronobis, W.; Von Lilienfeld, O.A.; Muller, K.R.; Tkatchenko, A. Machine learning predictions of molecular properties: Accurate many-body potentials and nonlocality in chemical space. J. Phys. Chem. Lett. 2015, 6, 2326–2331.
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36.
- Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. InChI—The worldwide chemical structure identifier standard. J. cheminform. J. Cheminform. 2013, 5, 1–9.
- Grethe, G.; Goodman, J.; Allen, C. International chemical identifier for chemical reactions. J. Cheminform. 2013, 5, O16.
- Elton, D.C.; Boukouvalas, Z.; Butrico, M.S.; Fuge, M.D.; Chung, P.W. Applying machine learning techniques to predict the properties of energetic materials. Sci. Rep. 2018, 8, 9059.
- Landrum, G. RDKit: Open-Source Cheminformatics Software. 2016. Available online: http://rdkit.org/ (accessed on 20 December 2020).
- Cxcalc, ChemAxon. Available online: https://www.chemaxon.com (accessed on 20 December 2020).
- Krenn, M.; Häse, F.; Nigam, A.; Friederich, P.; Aspuru-Guzik, A. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 2020, 1, 045024.
- Available online: https://aspuru.substack.com/p/molecular-graph-representations-and (accessed on 20 December 2020).
- Koichi, S.; Iwata, S.; Uno, T.; Koshino, H.; Satoh, H. Algorithm for advanced canonical coding of planar chemical structures that considers stereochemical and symmetric information. J. Chem. Inf. Model. 2007, 47, 1734–1746.
- O’Boyle, N.M. Towards a Universal SMILES representation—A standard method to generate canonical SMILES based on the InChI. J. Cheminform. 2012, 4, 1–14.
- Daylight Chemical Information Systems Inc. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html (accessed on 20 December 2020).
- O’Boyle, N.; Dalke, A. DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures. Chemrxiv 2018, 1–9.
- Maragakis, P.; Nisonoff, H.; Cole, B.; Shaw, D.E. A Deep-Learning View of Chemical Space Designed to Facilitate Drug Discovery. J. Chem. Inf. Model. 2020, 60, 4487–4496.
- Nigam, A.; Friederich, P.; Krenn, M.; Aspuru-Guzik, A. Augmenting Genetic Algorithms with Deep Neural Networks for Exploring the Chemical Space. arXiv 2020, arXiv:1909.11655.
- Gebauer, N.; Gastegger, M.; Schütt, K. Symmetry-adapted generation of 3d point sets for the targeted discovery of molecules. In Advances in Neural Information Processing Systems; Wallach, H., Larochelle, H., Beygelzimer, A., d’Alché-Buc, F., Fox, E., Garnett, R., Eds.; Curran Associates, Inc.: Vancouver, BC, Canada, 2019; Volume 32, pp. 7566–7578.
- Schütt, K.T.; Kessel, P.; Gastegger, M.; Nicoli, K.A.; Tkatchenko, A.; Müller, K.R. SchNetPack: A Deep Learning Toolbox For Atomistic Systems. J. Chem. Theory Comput. 2019, 15, 448–455.
- Minnich, A.J.; McLoughlin, K.; Tse, M.; Deng, J.; Weber, A.; Murad, N.; Madej, B.D.; Ramsundar, B.; Rush, T.; Calad-Thomson, S.; et al. AMPL: A Data-Driven Modeling Pipeline for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 1955–1968.
- St. John, P.C.; Phillips, C.; Kemper, T.W.; Wilson, A.N.; Guan, Y.; Crowley, M.F.; Nimlos, M.R.; Larsen, R.E. Message-passing neural networks for high-throughput polymer screening. J. Chem. Phys. 2019, 150, 234111.
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388.
- Göller, A.H.; Kuhnke, L.; Montanari, F.; Bonin, A.; Schneckener, S.; ter Laak, A.; Wichard, J.; Lobell, M.; Hillisch, A. Bayer’s in silico ADMET platform: A journey of machine learning over the past two decades. Drug Discov. Today 2020, 25, 1702–1709.
- Schütt, K.T.; Arbabzadah, F.; Chmiela, S.; Müller, K.R.; Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 13890.
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.J.; Tkatchenko, A.; Müller, K.R. SchNet—A deep learning architecture for molecules and materials. J. Chem. Phys. 2018, 148, 241722.
- Schütt, K.; Kindermans, P.J.; Sauceda Felix, H.E.; Chmiela, S.; Tkatchenko, A.; Müller, K.R. SchNet: A continuous-filter convolutional neural network for modeling quantum interactions. In Advances in Neural Information Processing Systems 30; Guyon, I., Luxburg, U.V., Bengio, S., Wallach, H., Fergus, R., Vishwanathan, S., Garnett, R., Eds.; Curran Associates, Inc.: Vancouver, BC, Canada, 2017; pp. 991–1001.
- Axelrod, S.; Gomez-Bombarelli, R. GEOM: Energy-annotated molecular conformations for property prediction and molecular generation. arXiv 2020, arXiv:2006.05531.
More
Information
Subjects:
Biophysics
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
29 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No