Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniel J. Klionsky | + 5357 word(s) | 5357 | 2021-11-15 09:33:13 | | | |
| 2 | Vivi Li | Meta information modification | 5357 | 2021-12-06 03:36:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Klionsky, D. Autophagy in Human Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/16731 (accessed on 25 July 2026).
Klionsky D. Autophagy in Human Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/16731. Accessed July 25, 2026.
Klionsky, Daniel. "Autophagy in Human Diseases" Encyclopedia, https://encyclopedia.pub/entry/16731 (accessed July 25, 2026).
Klionsky, D. (2021, December 03). Autophagy in Human Diseases. In Encyclopedia. https://encyclopedia.pub/entry/16731
Klionsky, Daniel. "Autophagy in Human Diseases." Encyclopedia. Web. 03 December, 2021.
Copy Citation
Autophagy, a process of cellular self-digestion, delivers intracellular components including superfluous and dysfunctional proteins and organelles to the lysosome for degradation and recycling and is important to maintain cellular homeostasis. In recent decades, autophagy has been found to help fight against a variety of human diseases, but, at the same time, autophagy can also promote the procession of certain pathologies, which makes the connection between autophagy and diseases complex but interesting.
autophagy
cancer
infection
metabolism
neurodegeneration
1. Introduction to Autophagy
Autophagy is a conserved process from yeast to human, in which parts of the cytoplasm are transported to the lysosome (the vacuole in fungi and plants) for degradation and recycling. Three primary types of autophagy have been characterized, macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) and the most prevalent form is macroautophagy (hereafter autophagy). The morphological hallmark of this form of autophagy is the formation of double-membrane vesicles, autophagosomes, which are the terminal product of phagophores. Following expansion and closure, the phagophore is now termed an autophagosome; this latter compartment ultimately fuses with the lysosome, releasing the cargo into the lumen of the degradative organelle where it is exposed to hydrolytic enzymes. The entire process of autophagy can be viewed as four steps: (1) initiation and nucleation of the phagophore; (2) expansion and closure of the phagophore to generate a completed autophagosome; (3) fusion with the lysosome; and (4) degradation and release/recycling of the autophagic cargos. Different autophagy-related (ATG) proteins are essential for each of these steps (Figure 1A).
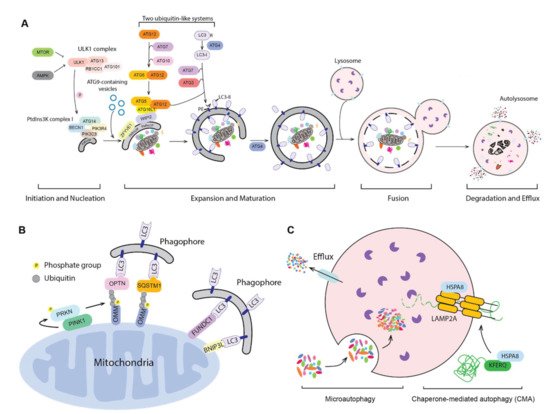
Figure 1. Three types of autophagy. (A) The mechanism of macroautophagy. Upon induction, the phagophore nucleates and expands to sequester cytoplasmic substrates randomly or selectively. Upon completion, the phagophore closes and forms a double-membrane autophagosome, which fuses with an endosome (not shown) and/or a lysosome; the autophagic cargos are degraded and the breakdown products are released back into the cytosol. The sequence shown for the LC3B C terminus is for the mouse protein. (B) PINK1- and PRKN-mediated and receptor-mediated mitophagy. Upon mitochondrial damage, PINK1 recruits and phosphorylates PRKN on the outer mitochondrial membrane (OMM). PRKN mediated the ubiquitination of OMM proteins, which then bind to autophagy receptors, targeting the mitochondria to a phagophore via interaction with an Atg8-family protein. Additionally, OMM proteins such as FUNDC1 and BNIP3L can serve as the mitophagy receptors. (C) Microautophagy and chaperone-mediated autophagy (CMA). During microautophagy, the lysosome membrane rearranges and forms a lumenal vesicle-containing autophagic substrates, which will be degraded within the lysosome. CMA starts from the recognition of a KFERQ-like motif in the substrate protein by HSPA8. HSPA8 facilitates the transport of substrates into the lysosome through the action of LAMP2A and lumenal HSPA8.
Initiation begins with the activation of the ULK1 complex, including ULK1, ATG13, RB1CC1 and ATG101 in response to MTORC1 and AMPK [1]. The activated ULK1 complex will then phosphorylate and activate the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, which contains PIK3C3/VPS34, PIK3R4/VPS15, BECN1, ATG14, NRBF2 and AMBRA1. The activated PIK3C3/VPS34 phosphorylates phosphatidylinositol (PtdIns) to produce a local pool of phosphatidylinositol-3-phosphate (PtdIns3P), which defines the region of phagophore initiation and recruits PtdIns3P effector proteins including WIPI2 and ZFYVE1/DFCP1 [2]. One model suggests that the phagophore is derived from a PtdIns3P-enriched ER region named the omegasome, followed by the recruitment of membrane sources for phagophore expansion, including ER exit sites [3], ER–mitochondria contact sites [4], the plasma membrane [5] and recycling endosomes [6], possibly via ATG9-containing vesicles, although the details of this process are still not fully understood. Another model, proposed by David Rubinsztein’s group, suggests that the phagophore is evolved from RAB11-positive recycling endosomes [7]. Even though the origin of the autophagosome membrane is still under debate, the hierarchical assembly of ATG proteins is well accepted.
Following initiation, the phagophore expands through the action of two ubiquitin-like protein systems [8]. In the first system, ATG12 is conjugated with ATG5 via the E1-like enzyme ATG7 and the E2-like enzyme ATG10. The second ubiquitin-like system starts with the proteolytic processing of Atg8-family proteins, including the MAP1LC3/LC3 and GABARAP subfamilies, by the ATG4 protease; this is followed by conjugation to phosphatidylethanolamine (PE), in a process termed lipidation, via ATG7, the E2-like enzyme ATG3 and the E3-like ATG12–ATG5-ATG16L1 complex [8]. ATG16L1 binds to WIPI2 directly, allowing the lipidation to take place at the phagophore membrane [9]. The expansion and sealing of the phagophore generates the double-membrane autophagosome; during this step the ATG proteins disassociate from the autophagosome outer membrane [10].
Finally, the autophagosome outer membrane fuses with an endosome to form an intermediate compartment termed an amphisome and/or with a lysosome to form an autolysosome. The autophagosome inner membrane, together with the sequestered cargo will be exposed to the lysosomal hydrolases and degraded. The resulting small molecules are released back into the cytosol through membrane permeases for reuse or as catabolic substrates.
2. Autophagy and Cancer
2.1. Autophagy and Tumor Suppression
In 1999, a study from Beth Levine’s lab demonstrated the role of BECN1 as a tumor suppressive factor, first linking autophagy with cancer [11]. Several subsequent studies have connected BECN1 with different types of cancer, such as hepatocellular carcinoma, gastric cancer and lymphoma [12][13][14]. The fact that the heterozygous disruption of BECN1 results in an increased frequency of spontaneous malignancies and decreased autophagy [15][16] indicates that autophagy functions as a tumor suppression mechanism. In line with this hypothesis, several BECN1-interacting proteins function as tumor suppressors. Frameshift and truncation mutations in UVRAG, an important BECN1 interactor during autophagosome and lysosome fusion [17], are found in cancer cells and also lead to reduced autophagy and increased tumorigenicity [18][19]. SH3GLB1/BIF1, which joins the BECN1 complex through UVRAG, is also a tumor suppressor; loss of SH3GLB1 inhibits autophagy while promoting tumorigenesis [20]. Another important protein for the BECN1-containing complex activity, AMBRA1 [21], is reported to act as a tumor suppressor via facilitating the degradation of the proto-oncogene MYC/c-Myc [22]. AMBRA1 also controls the cell cycle from the G1 to S phase through mediating D-type cyclin degradation, which reduces DNA replication stress, maintains genome integrity and therefore suppresses tumorigenesis [23].
Mutations of core ATG genes, including ATG2B, ATG5 and ATG9B, are also found in cancers, especially those with high microsatellite instability (MSI), which is characterized by the insertion or deletion in the repeated DNA sequence due to deficient DNA mismatch repair [24][25]. Additionally, the frameshift mutation of UVRAG mentioned above is also found in gastric carcinomas with MSI [18]. In addition to the core ATG genes, frameshift mutations in VPS33A, which functions during the fusion between autophagosomes and lysosomes [26], are also found in colorectal cancer cells with high MSI [27]. However, how these mutations affect autophagy and tumor progression has not been fully examined. In addition, mutation at an ATG5 splice site has been reported in a prostate cancer cell line, which prevents ATG12 conjugation and leads to the degradation of ATG12 and ATG16L1, thus inhibiting autophagy [28]. Of note, even though ATG genes have a more than 25% mutation rate in gastric and colorectal cancers with MSI [25], a study analyzing 11 different types of cancer supports the idea that core autophagy machinery is not highly targeted by single-nucleotide mutations [29]. This finding could reflect the necessity of an intact autophagy pathway for tumor growth. In line with this, the ablation of autophagy results in more benign diseases rather than invasive cancer [30][31][32][33], indicating that autophagy inhibits tumor initiation but is required for further progression, which will be discussed in the next subsection.
Most of the studies mentioned above focus on autophagy activity, but not its selectivity. Recently, by mapping mutations in cancers to LIR-motif-containing proteins, Han et al. found more than 200 potential LIR-motif associated mutations (LAMs) in 148 different proteins [34]. They further examined STBD1, a glycogen autophagy receptor, and found that it inhibits tumor growth and that the cancer-associated LAM inhibits its interaction with LC3 and abolishes its cancer-suppressing capability, drawing a connection between glycogen autophagy and tumor suppression [34]. Other proteins of interest were identified from this analysis. For instance, some core ATG genes were identified in the screen, including ATG4B, ATG2B, ATG5 and ATG9A, and the mutation in ATG4B decreases its interaction with LC3. How these mutations affect autophagy and how the potential change in autophagy is associated with cancer need further examination. Additionally, some proteins not in the core autophagy machinery are predicted to contains LAMs, such as BRAF, a defined oncogenic protein [35]. Can these proteins be the cargos or receptors of selective autophagy? Do these mutations in LIR motifs function as a way to escape autophagic degradation and result in cancer? The answer to these questions will shed more light on how autophagy suppresses tumors.
2.2. Autophagy and Tumor Promotion
Cancer cells often encounter a stressful environment lacking both nutrients and oxygen, especially in the interior region of a tumor. Because autophagy is an important pathway for cell survival under stress conditions, it is not surprising to see that autophagy supports tumor maintenance and growth. Elevated autophagy is discovered in many types of cancers, suggesting the important role of autophagy in tumor promotion [36][37]. In line with this observation, attenuated tumor growth has been found when autophagy is inhibited [32][33][38][39][40][41][42][43]. One important mechanism by which autophagy promotes tumor growth is to sustain metabolic plasticity [44]. Autophagy can recycle macromolecules and provide metabolic substrates to sustain energy homeostasis [45][46][47] and ablated autophagy in STK11/LKB1-deficient cells leads to insufficient amino acids for mitochondria energy production, excessive fatty acid oxidation and energy crisis [48]. Furthermore, blockage of autophagy also leads to the accumulation of ROS, resulting in DNA damage, which is another possible reason for impaired cancer cell growth after autophagy inhibition [38].
Importantly, even though tumor promotion through autophagy is discovered in many cancers, it depends on the genotype of the cancer cells, or more specifically, the expression of TP53/p53. In both breast cancer and non-small-cell lung cancer, inhibition of autophagy only impairs the growth of the cancer cells expressing TP53 [49][50], indicating that TP53 inhibits tumor growth when autophagy is inhibited. However, whether TP53 determines the consequence of autophagy inhibition is controversial when it comes to pancreatic ductal adenocarcinoma [31][51]. Overall, it seems unlikely that inhibiting autophagy can be used as a general therapeutic approach, but there may be patient-specific applications depending on the genotype of the cancer cells.
2.3. Autophagy and Tumor Metastasis
Metastasis is one of the hallmarks of cancer and causes most cancer-related deaths [52]. Increased expression of autophagy associated genes is correlated with a more aggressive and invasive phenotype [53][54], suggesting that autophagy primarily promotes tumor metastasis.
2.3.1. Autophagy and Cancer Cell Motility
The very first step of metastasis is to gain motility and invasiveness to allow migration from the primary tumor site. Cell migration is critical at the early stage of metastasis and turnover of focal adhesions (FAs) is important during this process [55]. FA turnover is promoted by autophagy, though degrading FA proteins such as PXN (paxillin) [56][57]. Additionally, a recent study shows that the deletion of RB1CC1 or ATG5 leads to different FA morphologies, but they both result in reduced cell motility [58]. However, during energy starvation, RB1CC1 is activated by ULK1 and inhibits focal adhesion kinase PTK2/FAK, leading to reduced cell motility and tumor metastasis [59]. Together, these studies suggest that energy-starvation-induced autophagy may inhibit cancer cell metastasis, but basal autophagy is necessary for cancer cells to gain motility and invasiveness.
NBR1 is also suggested to mediate the autophagy-dependent disassembly of FA proteins through its interaction with FA proteins and LC3 [60], suggesting the selective degradation of FA proteins. However, which FA protein or proteins are the cargo of NBR1-mediated selective autophagy is unknown. Additionally, because NBR1 contains a ubiquitin-binding domain, ubiquitination of FA proteins may participate in this process, but additional studies are needed to confirm this hypothesis. In a recent study, researchers from Jayanta Debanth’s lab reported that in breast cancer, autophagy inhibition promotes aberrant NBR1 accumulation, which induces the expression of basal epithelial markers and metastatic outgrowth [61]. Together, these studies indicate that autophagy may have distinct roles at different stages of metastasis, supporting cell matrix detachment in an early step, but suppressing metastatic outgrowth at later stages, and NBR1 seems to be a critical protein in both types of regulation. This idea is in line with some discoveries that metastases have a higher level of autophagy than the primary tumor cells, and early cancer metastases have the highest LC3 level [62]. However, many questions remain to be answered: What are the downstream targets of NBR1 when it promotes metastatic outgrowth? Overexpressing NBR1 promotes SQSTM1 accumulation and phosphorylation in liquid-like bodies [63] and SQSTM1-mediated activation of NFE2L2/NRF2 provides hepatocellular carcinoma cells proliferation potency [64]. In addition, aberrant regulation of NFE2L2 is correlated with high-level resistance to anticancer drugs [65]. Therefore, NBR1 accumulation by autophagy inhibition may help tumor metastases outgrowth through SQSTM1 and NFE2L2, but this still needs further investigation. Additionally, how NBR1 is regulated and plays different roles in cancer cell motility and metastases outgrowth and whether NBR1 is involved in other cancers need further investigation.
Epithelial-mesenchymal transition (EMT), a pro-metastatic process wherein epithelial cells gain mobility and invasiveness to become mesenchymal stem cells, is also regulated by autophagy [66]. Autophagy, on the one hand, promotes EMT [67][68][69][70]; however, how this happens remains controversial. Some studies indicate that autophagy regulates EMT in a TGFB-dependent manner [67], but others point out that TGFB-induced EMT partially depends on autophagy [71]. Therefore, which pathway is upstream remains unclear. Additionally, because TGFB can activate autophagy [72], whether there is a positive loop between these two factors to promote EMT is worth examining. On the other hand, autophagy could inhibit EMT via selective degradation of the transcriptional repressor SNAI1/Snail and inhibiting SQSTM1-dependent stabilization of the transcription factor TWIST1 [73][74][75]. These discrepancies in how autophagy regulates EMT, whether in the same direction but via different mechanisms or even in opposite directions, indicate the importance of figuring out if autophagy is directly or indirectly linked to EMT. If directly, does it act in parallel with the known EMT signaling pathways? If indirectly, apart from TGFB, are there interactions between autophagy and EMT pathways that contribute to cancer cell EMT? This is a pertinent questions because, besides the TGFB-SMAD pathway, other types of cell signaling, including PI3K-AKT, MAPK, and RHO GTPase cascades [76] are also key mediators to activate the EMT and they all have close interactions with autophagy [77]; thus, it is possible that autophagy either acts in parallel with one or more of these pathways or they function together to form a network regulating cancer cell EMT.
2.4. Autophagy and Cancer Stem Cells
Cancer stem cells (CSC) are a subpopulation of cancer cells, which are similar to normal stem cells, but proposed to be critical for tumor metastasis because of their high mobility and self-renewal ability [78]. Autophagy is upregulated in a variety of CSCs and found to be important for CSC survival and maintaining stemness, which encompasses the fundamental properties of stem cells such as self-renewal and generating daughter cells [79][80][81]. Previously, IL17B was shown to be overexpressed in breast cancer tissue and inversely correlate with breast cancer patient survival rate [82], and a recent study indicates that IL17B induces autophagosome formation in gastric cancer CSCs; in contrast, autophagy inhibition through ATG7 deletion inhibits IL17B-induced self-renewal, which draws a connection between autophagy and CSC maintenance [83]. The ability of autophagy to maintain CSCs may provide an explanation as to why some ATG genes, such as LC3B, GABARAP and ATG5, have been found to correlate with poor prognosis [84][85].
Autophagy supports CSC stemness through several downstream pathways. First, in breast cancer, autophagy supports stemness through inducing the secretion of IL6 [86], which is important for stemness maintenance [87]. Second, EGFR-STAT3 and TGFB-SMAD signaling pathways are found to act downstream of autophagy to sustain CSC stemness [88]. Third, in gastric CSCs, a higher expression level of FOXA2 is sustained by autophagy [89], which promotes cell proliferation and maintain CSC stemness [90], and overexpressing FOXA2 partially rescues the decreased self-renewal ability when autophagy is inhibited [89]. Finally, autophagy can augment cell stemness through degrading ubiquitinated TP53 [85][91]. All the studies mentioned above indicate that autophagy positively regulates CSC. However, this observation does not mean that continually higher autophagy activity is better for CSCs. A study from Shashi Gujar’s lab demonstrates that autophagy promotion and suppression both result in a decrease in pluripotency and an increased differentiation or senescence of CSCs [92], indicating that a proper autophagy level is essential to sustain the stemness of CSCs.
2.5. Autophagy and Dormant Cancer Cells
One reason tumors are difficult to treat is the existence of dormant cancer cells, which refers to some cancer cells that have arrested growth but are able to retain proliferative capacity and lead to subsequent tumor growth [93][94]. Even though dormant cells have similarities to CSCs such as drug resistance, fundamental differences exist. CSCs are considered as “slow cycling cells” whereas dormant cancer cells undergo cell cycle arrest. In addition, CSCs express stemness marker genes and are at the apex of the differentiation hierarchy. However, dormant and activated cancer cells are at the same differentiation stage and the switch between dormancy and activation is reversible [95].
Many independent studies show that under different dormancy induction conditions, cancer cells show a higher autophagy activity [96], suggesting that autophagy may be critical to maintain the survival of these dormant cells. A majority of ovarian cancer patients develop tumor recurrence, possibly due to the existence of dormant cancer cells [97], and therefore, ovarian cancer becomes a major model to study cancer dormancy. DIRAS3/ARHI usually has a lower expression level in ovarian cancer cells, but the re-expression of DIRAS3 induces autophagy and these cells keep dormant when they grow in a mouse model. The inhibition of autophagy through chloroquine (CQ) treatment leads to a reduced regrowth of these dormant cells in the mouse model, indicating that DIRAS3-induced autophagy is critical for the survival of dormant cancer cells [98][99]. Another study shows that AKT inhibition induces dormancy-like ovarian cancer cells. Under this condition, autophagy activity is increased, and autophagy inhibition will reduce cell viability [100]. Interestingly, DIRAS3-induced autophagy seems to play a different role in vitro and in vivo. As mentioned previously, expressing DIRAS3 in xenograft leads to dormant cancer cells, whereas in cell culture, autophagy induction through DIRAS3 leads to cell death [101]. This difference may come from a more complicated in vivo system than the in vitro cell culture and the interaction between the cancer cell and its microenvironment may contribute to the choice between apoptosis or quiescence. Beyond ovarian cancer, cancer cell dormancy promotion by autophagy is seen in many other cancer types as well. Dormant breast CSCs are highly autophagic [102], which supports cell survival during dormancy [103]. In glioblastoma, autophagy reprograms cancer cell metabolism and promotes cancer cell quiescence [104].
Inhibition of autophagy facilitates cancer cell escape from the dormant state [102][105]. PFKFB3, which may promote cancer cell metastasis [106], could be a key factor in this process. PFKFB3 is an autophagy substrate and an elevated level of PFKFB3 when autophagy is impaired may explain the induced tumor recurrence [102]. Recently, MIR27A was reported to ameliorate chemoresistance of breast cancer cells and, at the same time, inhibit autophagy [107], further suggesting that impairment of autophagy leads to cancer cell emergence from the dormant state.
Whether autophagy could be a target to deal with dormant cancer cells having high drug resistance needs a more careful examination. Is awakening dormant tumors via inhibiting autophagy a good way to make them more sensitive to chemotherapy and preventing tumor recurrence? A recent study suggests that treating DIRAS3-overexpressing ovarian dormant cancer cells with crizotinib further increases autophagy and induces apoptosis, and treating mice carrying DIRAS3-expressing ovarian cancer cells with crizotinib prolongs life span [108], even though, as mentioned above, DIRAS3-indcued autophagy supports dormant cancer cell survival. These findings suggest that autophagy may have dual roles in sustaining tumor dormancy and that inducing autophagy to an improperly high level may also become a way to eliminate dormant cancer cells.
3. Autophagy and Neurodegenerative Diseases
One hallmark of neurodegenerative diseases is the abnormal accumulation of certain neuroproteins. Because autophagy is critical for the degradation of protein aggregates and maintaining cellular homeostasis, it is not surprising to see that autophagy has a close connection with neurodegeneration: autophagy is responsible for the clearance of accumulated proteins, and this role is particularly important in non-dividing cells. In this section, we will discuss the role of autophagy in Parkinson, Alzheimer, and Huntington diseases.
3.1. Parkinson Disease
PD is characterized by the progressive loss of dopaminergic neurons of the substantia nigra, which is accompanied by the accumulation of SNCA/α-synuclein in the form of Lewy bodies and Lewy neurites [109]. From genome-wide association studies (GWAS), great advances have been made in recent decades with the identification of monogenetic causes of PD, including mutations in SNCA, LRRK2, PRKN, and PINK1 [110][111].
SNCA is a substrate of CMA [112], and, consistent with this fact, boosting CMA decreases SNCA levels and protects cells from wild-type SNCA-induced neurotoxicity [113]. The SNCA accumulation and neurotoxicity in PD patients may result from two factors related to CMA. First, PD-associated mutant SNCA, A53T and A30P, are degraded by CMA less efficiently because they bind to the lysosome but cannot be translocated into the lysosomal lumen, which, at the same time, inhibits the degradation of other CMA cargos and increases cell toxicity [114][115]. Second, the expression level of essential CMA proteins, such as LAMP2A and HSPA8, decreases significantly in PD patient brains [116][117][118]. In addition, the PD-associated UCHL1I93M mutation facilitates interaction with LAMP2A, thus inhibiting CMA [119].
Besides CMA, SNCA is degraded through macroautophagy in neuronal cells [112] and particularly, via selective-autophagy mediated by SQSTM1 as the receptor in microglia [120]. At the same time, SNCA regulates autophagy. Overexpression of SNCA inhibits autophagy via RAB1, which further leads to the mislocalization of ATG9 [121]. Overexpression of PD-associated mutant SNCAE46K impairs autophagosome formation through the inactivation of the MAPK8/JNK1-BCL2 pathway [122]. One recent study, expressing human SNCA in Drosophila, found that SNCA impairs macroautophagy through stabilizing the actin cytoskeleton, which inhibits the fusion between lysosomes and autophagosomes [123]. Even though SNCA is a target of autophagy, SNCA aggregates are not easily degraded by autophagy and inhibit this process by impairing autophagosome clearance [124]. Autophagy is not only responsible for the degradation of SNCA, but also affects its cell-to-cell transmission [125]. Several studies indicate that the blockage of autophagy induces SNCA secretion through exosomes [126][127][128], which reduces cell death, but creates a microenvironment with an inflammatory and neurotoxic response [128]. Additionally, the secreted SNCA will be taken up by other neurons and act as a seed for aggregation in the recipient cells [129].
LRRK2 mutations are one of the most common causes of PD. In most cases of LRRK2-associated PD, the protein has the G2019S mutation and the cells display the SNCA aggregates as Lewy bodies and undergo cell death [130]. Similar to SNCA, LRRK2 is another substrate of CMA, but the LRRK2G2019S mutant is again difficult to degrade and inhibits CMA, which underlies the toxicity in PD by compromising the CMA-mediated degradation of SNCA [131][132]. Besides, LRRK2R1441G, which leads to age-dependent SNCA accumulation, also inhibits CMA [132]. LRRK2 regulates macroautophagy as well, but the role remains undetermined. Many studies, involving LRRK2 kinase inhibitor and LRRK2G2019S, which has higher kinase activity, demonstrate that LRRK2 inhibits autophagy [133][134]. In contrast, some studies indicate that LRRK2 may promote autophagy through the activation of the MAP2K/MEK-MAPK/JNK-MAPK/ERK pathway [135] and it is reported that age-dependent dopaminergic neurodegeneration and autophagy impairment occur in lrrk1 lrrk2 double-knockout mice [136]. Further studies should integrate these relevant findings and draw a more complete model of how LRRK2 affects autophagy, which will be of great significance in designing autophagy-targeting PD therapy.
Mitochondria dysfunction has long been recognized as the initiating factor in dopaminergic neuronal loss [137]. Of note, mutations in PINK1 and PRKN, two critical proteins in mitophagy, are highly associated with PD [138]. Interestingly, PD-associated PINK1 mutations are clustered in the kinase domain [138] and several mutations such as G309D, L347P and W437X have a compromised interaction with PRKN, thus inhibiting mitophagy execution [139]. In addition to the mutations in these two proteins, studies focused on other PD-associated proteins also shed light on the importance of mitophagy in PD. Pathogenic SNCA impairs mitochondrial function via binding to OMM proteins such as TOMM20, which impairs protein import to mitochondria [140][141][142], or decreasing the mitochondrial SIRT3 level [143]. As mentioned above, mitophagy is responsible for impaired mitochondria degradation and a study expressing SNCA in yeast shows that Sir2-mediated mitophagy is induced and the selective degradation of mitochondria is responsible for the SNCA toxicity [144]. However, in neurons or in in vivo models, how SNCA-mediated mitochondrial damage is related to or affects mitophagy is unknown. Compared with SNCA, there are more studies about LRRK2 and mitophagy. It is found that the PD-associated LRRK2G2019S mutation inhibits mitophagy by affecting mitochondria motility [145], inhibiting mitochondrial fission [146], and phosphorylating RAB10 to inhibit its mitochondrial accumulation and interaction with OPTN [147].
Here, we focus on SNCA, LRRK2, PINK1 and PRKN, summarizing how they interact with autophagy. Other proteins with PD-associated mutations, such as VPS35, VPS13C and FBXO7, have been suggested to play a role in autophagy (Table 1).
Table 1. PD-associated genes from GWAS and their connection with autophagy (except SNCA, LRKK2, PINK1 and PRKN).
| Gene Name | Description | Reference |
|---|---|---|
| GBA | Loss of GBA function impairs autophagy via PPP2/PP2A inactivation. PD-associated mutation L444P heterozygote impairs autophagy, mitochondria priming and autophagy-lysosome degradation. |
[148][149] |
| VPS13C | Deletion of VPS13C is correlated with impaired mitochondrial morphology and upregulate PINK1-PRKN-dependent mitophagy, but the study does not show the connection between PD-associated mutations with mitophagy. | [150] |
| VPS35 | VPS35D620N causes autosomal-dominant Parkinson disease. VPS35D620N has a reduced affinity for WASH and impairs ATG9A trafficking and localization, thus compromising autophagosome formation. |
[151] |
| VPS35D620N impairs endosome-to-Golgi retrieval of LAMP2A and accelerates LAMP2A degradation, thus inhibiting SNCA degradation through CMA. | [152] | |
| VPS35D620N hampers PINK1 and PRKN recruitment to mitochondria thus impairing mitophagy. | [153] | |
| PARK7 | PARK7 knockdown impairs autophagy and the SNCA uptake and degradation in microglia. | [154] |
| PARK7 deficiency downregulates HSPA8 expression level and accelerates the degradation of LAMP2A, inhibiting SNCA degradation through CMA. | [155] | |
| Park7 may function in mitophagy because it is important for proper mitochondria function and Park7 upregulation can rescue the phenotype in pink1 mutant Drosophila. | [156] | |
| SREBF1 | SREBF1 knockdown inhibits PRKN translocation to mitochondria, thus inhibiting mitophagy. | [157] |
| FBXO7 | FBXO7T22M inhibits its interaction with PRKN and impairs PRKN translocation to mitochondria. | [158] |
| FBXO7R378G mutation impairs ubiquitination of MFN1. | ||
| FBXO7R498X truncation inhibits PRKN recruitment to mitochondria. | ||
| T22M, R378G and R498X mutations aggravate aggregation of FBXO7 in mitochondria, which may inhibit mitophagy. | [159] | |
| TMEM175 | TMEM175 deficiency leads to the impaired autophagosome degradation in the lysosome. | [160] |
| TMEM175M393T shows similar autophagosome clearance phenotype as a knockout. | [161] |
GWAS provides us with invaluable information to study the connection between PD and autophagy and identify therapeutic targets. However, with 90 variants nominated as PD-related factors [110], how to study them, particularly how they are related to autophagy, needs further consideration. First, some PD-associated genes are only studied by knockout instead of using the PD-associated mutated form (such as VPS13C [150]). Even though studies of mutant proteins may lead to the concern that the point mutation may not be sufficient to result in either an autophagy or pathological phenotype, further studies focusing on the mutation may shed light on a more detailed mechanism of PD and autophagy. Second, some PD-associated genes discovered through GWAS studies may affect autophagy, but few studies delve into them with regard to mechanism. For instance, several genes, such as CHCHD2 [162] and ATP13A2 [163][164], are critical for mitochondrial quality, but whether they have any connection with mitophagy remains unclear. Third, controversial data exist, which may have resulted from the use of different cells lines, and the phenotype at the cellular level is sometimes different from that at the behavioral level [165]. Therefore, based on the goal of specific studies, the model used to study these genes and what marker(s)/phenotype(s) should be used as an indication of PD need consideration as well.
3.2. Alzheimer Disease
Alzheimer disease (AD) is a progressive neurodegenerative disease characterized by cognitive impairment and loss of memory. AD patients usually show the accumulation of misfolded proteins such as amyloid-β (Aβ) and hyperphosphorylated MAPT (microtubule associated protein tau) [166].
Several lines of evidence indicate deficient autophagy in AD patients, which include the decreased level of autophagy-related genes, including BECN1 [167], ATG5 and LC3B [168], and the accumulation of autophagosomes [169]. More importantly, the accumulation of autophagosomes correlates with AD pathology [170], which further indicates the importance of understanding the connections between autophagy and AD. Besides these direct lines of evidence, AD-associated mutation in PSEN1 disrupt autophagy [171][172]. A decreasing level of PICALM, which occurs in AD, inhibits autophagy and exacerbates AD pathology [173][174][175]. However, some studies report an increase in autophagy when cells are treated with Aβ [176]. This discrepancy could at least in part be a consequence of the AD stage. In 2016, Bordi et al. carried out a comprehensive analysis at different stages of AD, finding an upregulation of autophagy-related genes at the early stage, but an impeded autophagy flux at the late stage [177]. The mechanism of this change is not clear, possibly because autophagy is induced at the early stage to degrade the protein aggregates. However, at the later stage, autophagy or lysosome clearance ability becomes inhibited by the accumulation of abnormal proteins.
The relationship between Aβ and autophagy is complicated. First, Aβ is degraded through autophagy, and several studies show a decreased Aβ level in cells and improved cognitive ability in an AD mouse model when autophagy is induced [178][179][180][181]. Second, Aβ may also be generated inside autophagosomes because both APP (amyloid beta precursor protein) and PSEN1, an enzyme involved in the cleavage of APP to form Aβ, are found within the autophagosome [169]. Third, one study reported that the secretion of Aβ to the extracellular space, where plaque forms, depends on autophagy in neurons [182]. On the contrary, a recent study indicates that MTORC1 inhibition reduces amyloid secretion due to the upregulation of autophagy [183]. Interestingly, the activation of AMPK does not induce autophagy in neurons, and different AMPK activators results in differential regulation of Aβ secretion, either increasing or reducing, which indicates a complex role of AMPK in Aβ secretion independent from autophagy [183].
APP has a KFERQ motif, which is typically associated with CMA. However, the deletion of this motif in APP does not abolish its interaction with HSPA8, but conversely, increases the interaction [184]. The authors raise the possibility that the KFERQ motif may be used to bind to AP2, which is an autophagy adaptor, because the AP2 recognition sequence is part of KFERQ; deletion of the KFERQ motif impairs AP2-dependent targeting to the lysosome. Further studies should investigate the binding partner of the KFERQ motif other than HSPA8; this may lead to the identification of new functions of this motif other than acting as the CMA signal and shed more light on AD pathology.
The other hallmark protein in AD, MAPT, is a substrate of macroautophagy, CMA and microautophagy, but some AD-associated MAPT mutations cannot be cleared efficiently by autophagy [185][186]. Consistently, activation of autophagy through inhibiting MTORC1 helps with prolonged clearance of MAPT [187]. CMA is downregulated in AD patient brains [188]; CMA upregulation improves the disease phenotype that results from MAPT or combined MAPT and Aβ pathologies, and inhibition of CMA accelerates the AD pathology in a mouse model [188].
Together with Aβ and MAPT, compromised mitochondria accumulation is another hallmark of AD. Although it is unclear whether the mitochondrial dysfunction is a cause or a consequence of Aβ and phosphorylated MAPT accumulation [189], it indicates that quality control of mitochondria is impaired in AD neurons. Deficient mitophagy has been discovered in AD patient brain and patient stem-cell derived neurons [190]. The compromised mitophagy in AD may resulted from the following. First, in AD patient brains, PINK1-PRKN-dependent mitophagy is enhanced with Aβ accumulation, but it is followed by a progressively depleted PRKN [191][192], suggesting that mitophagy is induced at the early stage of AD, but finally shows an inadequate capacity compared with the huge number of damaged mitochondria. Additionally, higher levels of Δ1 PINK1, the main cleaved product of PINK1, is found in AD patient brain, which inhibits PRKN translocation to mitochondria and impairs mitophagy [192]. Second, emerging data are showing that Aβ and phosphorylated MAPT interfere in the mitophagy pathway and MAPT impairs PRKN translocation to mitochondria [193][194][195]. Overall, these studies demonstrate that compromised mitophagy and abnormal mitochondrial dynamics contribute to AD pathogenesis.
References
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137.
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278.
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell. 2013, 24, 2918–2931.
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393.
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell. Biol. 2010, 12, 747–757.
- Puri, C.; Renna, M.; Bento, C.F.; Moreau, K.; Rubinsztein, D.C. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013, 154, 1285–1299.
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell. 2018, 45, 114–131.
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025.
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell. 2014, 55, 238–252.
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566.
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676.
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175.
- Zhou, W.H.; Tang, F.; Xu, J.; Wu, X.; Yang, S.B.; Feng, Z.Y.; Ding, Y.G.; Wan, X.B.; Guan, Z.; Li, H.G.; et al. Low expression of Beclin 1, associated with high Bcl-xL, predicts a malignant phenotype and poor prognosis of gastric cancer. Autophagy 2012, 8, 389–400.
- Nicotra, G.; Mercalli, F.; Peracchio, C.; Castino, R.; Follo, C.; Valente, G.; Isidoro, C. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod. Pathol. 2010, 23, 937–950.
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820.
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082.
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149.
- Kim, M.S.; Jeong, E.G.; Ahn, C.H.; Kim, S.S.; Lee, S.H.; Yoo, N.J. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum. Pathol. 2008, 39, 1059–1063.
- He, S.; Zhao, Z.; Yang, Y.; O’Connell, D.; Zhang, X.; Oh, S.; Ma, B.; Lee, J.H.; Zhang, T.; Varghese, B.; et al. Truncating mutation in the autophagy gene UVRAG confers oncogenic properties and chemosensitivity in colorectal cancers. Nat. Commun. 2015, 6, 7839.
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151.
- Fimia, G.M.; Di Bartolomeo, S.; Piacentini, M.; Cecconi, F. Unleashing the Ambra1-Beclin 1 complex from dynein chains: Ulk1 sets Ambra1 free to induce autophagy. Autophagy 2011, 7, 115–117.
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 706.
- Maiani, E.; Milletti, G.; Nazio, F.; Holdgaard, S.G.; Bartkova, J.; Rizza, S.; Cianfanelli, V.; Lorente, M.; Simoneschi, D.; Di Marco, M.; et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature 2021, 592, 799–803.
- Velho, S.; Fernandes, M.S.; Leite, M.; Figueiredo, C.; Seruca, R. Causes and consequences of microsatellite instability in gastric carcinogenesis. World J. Gastroenterol. 2014, 20, 16433–16442.
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706.
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell. 2014, 25, 1327–1337.
- An, C.H.; Kim, Y.R.; Kim, H.S.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Frameshift mutations of vacuolar protein sorting genes in gastric and colorectal cancers with microsatellite instability. Hum. Pathol. 2012, 43, 40–47.
- Wible, D.J.; Chao, H.P.; Tang, D.G.; Bratton, S.B. Cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12-ATG5-ATG16L1 complex assembly and autophagy. Cell Discov. 2019, 5, 42.
- Lebovitz, C.B.; Robertson, A.G.; Goya, R.; Jones, S.J.; Morin, R.D.; Marra, M.A.; Gorski, S.M. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015, 11, 1668–1687.
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800.
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913.
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461.
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927.
- Han, Z.; Zhang, W.; Ning, W.; Wang, C.; Deng, W.; Li, Z.; Shang, Z.; Shen, X.; Liu, X.; Baba, O.; et al. Model-based analysis uncovers mutations altering autophagy selectivity in human cancer. Nat. Commun. 2021, 12, 3258.
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197.
- Yoshioka, A.; Miyata, H.; Doki, Y.; Yamasaki, M.; Sohma, I.; Gotoh, K.; Takiguchi, S.; Fujiwara, Y.; Uchiyama, Y.; Monden, M. LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int. J. Oncol. 2008, 33, 461–468.
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365.
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729.
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527.
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216.
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439.
- Lévy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; Lemarchand, J.; Durand, A.; Crain, A.M.; et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 2015, 17, 1062–1073.
- Santanam, U.; Banach-Petrosky, W.; Abate-Shen, C.; Shen, M.M.; White, E.; DiPaola, R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016, 30, 399–407.
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619.
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470.
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285.
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 2016, 30, 1704–1717.
- Bhatt, V.; Khayati, K.; Hu, Z.S.; Lee, A.; Kamran, W.; Su, X.; Guo, J.Y. Autophagy modulates lipid metabolism to maintain metabolic flexibility for. Genes Dev. 2019, 33, 150–165.
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-Colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907.
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056.
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28.
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 2012, 18, 370–379.
- Zhao, H.; Yang, M.; Zhao, J.; Wang, J.; Zhang, Y.; Zhang, Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med. Oncol. 2013, 30, 475.
- Nagano, M.; Hoshino, D.; Koshikawa, N.; Akizawa, T.; Seiki, M. Turnover of focal adhesions and cancer cell migration. Int. J. Cell. Biol. 2012, 2012, 310616.
- Chang, C.H.; Bijian, K.; Qiu, D.; Su, J.; Saad, A.; Dahabieh, M.S.; Miller, W.H.; Alaoui-Jamali, M.A. Endosomal sorting and c-Cbl targeting of paxillin to autophagosomes regulate cell-matrix adhesion turnover in human breast cancer cells. Oncotarget 2017, 8, 31199–31214.
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell. Rep. 2016, 15, 1660–1672.
- Assar, E.A.; Tumbarello, D.A. Loss of the Essential Autophagy Regulators FIP200 or Atg5 Leads to Distinct Effects on Focal Adhesion Composition and Organization. Front. Cell. Dev. Biol. 2020, 8, 733.
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Invest. 2013, 123, 2907–2920.
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR1 enables autophagy-dependent focal adhesion turnover. J. Cell. Biol. 2016, 212, 577–590.
- Marsh, T.; Kenific, C.M.; Suresh, D.; Gonzalez, H.; Shamir, E.R.; Mei, W.; Tankka, A.; Leidal, A.M.; Kalavacherla, S.; Woo, K.; et al. Autophagic Degradation of NBR1 Restricts Metastatic Outgrowth during Mammary Tumor Progression. Dev. Cell. 2020, 52, 591–604.
- Peng, Y.F.; Shi, Y.H.; Shen, Y.H.; Ding, Z.B.; Ke, A.W.; Zhou, J.; Qiu, S.J.; Fan, J. Promoting colonization in metastatic HCC cells by modulation of autophagy. PLoS ONE 2013, 8, e74407.
- Sanchez-Martin, P.; Sou, Y.S.; Kageyama, S.; Koike, M.; Waguri, S.; Komatsu, M. NBR1-mediated p62-liquid droplets enhance the Keap1-Nrf2 system. EMBO Rep. 2020, 21, e48902.
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030.
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85.
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292.
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351.
- Kim, Y.H.; Baek, S.H.; Kim, E.K.; Ha, J.M.; Jin, S.Y.; Lee, H.S.; Ha, H.K.; Song, S.H.; Kim, S.J.; Shin, H.K.; et al. Uncoordinated 51-like kinase 2 signaling pathway regulates epithelial-mesenchymal transition in A549 lung cancer cells. FEBS Lett. 2016, 590, 1365–1374.
- Zhang, W.; Yuan, W.; Song, J.; Wang, S.; Gu, X. LncRNA CPS1-IT1 suppresses EMT and metastasis of colorectal cancer by inhibiting hypoxia-induced autophagy through inactivation of HIF-1α. Biochimie 2018, 144, 21–27.
- Ren, T.; Zheng, B.; Huang, Y.; Wang, S.; Bao, X.; Liu, K.; Guo, W. Osteosarcoma cell intrinsic PD-L2 signals promote invasion and metastasis via the RhoA-ROCK-LIMK2 and autophagy pathways. Cell. Death Dis. 2019, 10, 261.
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768.
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852.
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.W. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 2012, 72, 3238–3250.
- Grassi, G.; Di Caprio, G.; Santangelo, L.; Fimia, G.M.; Cozzolino, A.M.; Komatsu, M.; Ippolito, G.; Tripodi, M.; Alonzi, T. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis. 2015, 6, e1880.
- Qiang, L.; Zhao, B.; Ming, M.; Wang, N.; He, T.C.; Hwang, S.; Thorburn, A.; He, Y.Y. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc. Natl. Acad. Sci. USA 2014, 111, 9241–9246.
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234.
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 14, 1110–1128.
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564.
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Deloménie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272.
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell. Death Dis. 2018, 9, 841.
- Zhang, D.; Zhao, Q.; Sun, H.; Yin, L.; Wu, J.; Xu, J.; He, T.; Yang, C.; Liang, C. Defective autophagy leads to the suppression of stem-like features of CD271. J. Biomed. Sci. 2016, 23, 82.
- Huang, C.K.; Yang, C.Y.; Jeng, Y.M.; Chen, C.L.; Wu, H.H.; Chang, Y.C.; Ma, C.; Kuo, W.H.; Chang, K.J.; Shew, J.Y.; et al. Autocrine/paracrine mechanism of interleukin-17B receptor promotes breast tumorigenesis through NF-κB-mediated antiapoptotic pathway. Oncogene 2014, 33, 2968–2977.
- Bie, Q.; Song, H.; Chen, X.; Yang, X.; Shi, S.; Zhang, L.; Zhao, R.; Wei, L.; Zhang, B.; Xiong, H. IL-17B/IL-17RB signaling cascade contributes to self-renewal and tumorigenesis of cancer stem cells by regulating Beclin-1 ubiquitination. Oncogene 2021, 40, 2200–2216.
- Bortnik, S.; Tessier-Cloutier, B.; Leung, S.; Xu, J.; Asleh, K.; Burugu, S.; Magrill, J.; Greening, K.; Derakhshan, F.; Yip, S.; et al. Differential expression and prognostic relevance of autophagy-related markers ATG4B, GABARAP, and LC3B in breast cancer. Breast Cancer Res. Treat. 2020, 183, 525–547.
- Wang, J.; Liu, D.; Sun, Z.; Ye, T.; Li, J.; Zeng, B.; Zhao, Q.; Rosie Xing, H. Autophagy augments the self-renewal of lung cancer stem cells by the degradation of ubiquitinated p53. Cell. Death Dis. 2021, 12, 98.
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658.
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402.
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.L. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfβ/Smad Signaling. Cancer Res. 2016, 76, 3397–3410.
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171.
- Perez-Balaguer, A.; Ortiz-Martínez, F.; García-Martínez, A.; Pomares-Navarro, C.; Lerma, E.; Peiró, G. FOXA2 mRNA expression is associated with relapse in patients with Triple-Negative/Basal-like breast carcinoma. Breast Cancer Res. Treat. 2015, 153, 465–474.
- Ghatak, D.; Das Ghosh, D.; Roychoudhury, S. Cancer Stemness: p53 at the Wheel. Front Oncol. 2020, 10, 604124.
- Sharif, T.; Martell, E.; Dai, C.; Kennedy, B.E.; Murphy, P.; Clements, D.R.; Kim, Y.; Lee, P.W.; Gujar, S.A. Autophagic homeostasis is required for the pluripotency of cancer stem cells. Autophagy 2017, 13, 264–284.
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622.
- Yeh, A.C.; Ramaswamy, S. Mechanisms of Cancer Cell Dormancy--Another Hallmark of Cancer? Cancer Res. 2015, 75, 5014–5022.
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411.
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and Cancer Dormancy. Front. Oncol. 2021, 11, 627023.
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156.
- Lu, Z.; Baquero, M.T.; Yang, H.; Yang, M.; Reger, A.S.; Kim, C.; Levine, D.A.; Clarke, C.H.; Liao, W.S.; Bast, R.C. DIRAS3 regulates the autophagosome initiation complex in dormant ovarian cancer cells. Autophagy 2014, 10, 1071–1092.
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929.
- Correa, R.J.; Valdes, Y.R.; Peart, T.M.; Fazio, E.N.; Bertrand, M.; McGee, J.; Préfontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Combination of AKT inhibition with autophagy blockade effectively reduces ascites-derived ovarian cancer cell viability. Carcinogenesis 2014, 35, 1951–1961.
- Washington, M.N.; Suh, G.; Orozco, A.F.; Sutton, M.N.; Yang, H.; Wang, Y.; Mao, W.; Millward, S.; Ornelas, A.; Atkinson, N.; et al. ARHI (DIRAS3)-mediated autophagy-associated cell death enhances chemosensitivity to cisplatin in ovarian cancer cell lines and xenografts. Cell. Death Dis. 2015, 6, e1836.
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10, 3668.
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944.
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J.; et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell. Death Dis. 2018, 9, 213.
- Aqbi, H.F.; Tyutyunyk-Massey, L.; Keim, R.C.; Butler, S.E.; Thekkudan, T.; Joshi, S.; Smith, T.M.; Bandyopadhyay, D.; Idowu, M.O.; Bear, H.D.; et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget 2018, 9, 22113–22122.
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044.
- Ueda, S.; Takanashi, M.; Sudo, K.; Kanekura, K.; Kuroda, M. miR-27a ameliorates chemoresistance of breast cancer cells by disruption of reactive oxygen species homeostasis and impairment of autophagy. Lab. Investig. 2020, 100, 863–873.
- Blessing, A.M.; Santiago-O’Farrill, J.M.; Mao, W.; Pang, L.; Ning, J.; Pak, D.; Bollu, L.R.; Rask, P.; Iles, L.; Yang, H.; et al. Elimination of dormant, autophagic ovarian cancer cells and xenografts through enhanced sensitivity to anaplastic lymphoma kinase inhibition. Cancer 2020, 126, 3579–3592.
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease - lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29.
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. Neurol. 2019, 18, 1091–1102.
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516.
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556.
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146.
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295.
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515.
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472.
- Wu, G.; Wang, X.; Feng, X.; Zhang, A.; Li, J.; Gu, K.; Huang, J.; Pang, S.; Dong, H.; Gao, H.; et al. Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic Parkinson’s disease. Brain Res. 2011, 1394, 105–111.
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647.
- Kabuta, T.; Furuta, A.; Aoki, S.; Furuta, K.; Wada, K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem. 2008, 283, 23731–23738.
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386.
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell. Biol. 2010, 190, 1023–1037.
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant α-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701.
- Sarkar, S.; Olsen, A.L.; Sygnecka, K.; Lohr, K.M.; Feany, M.B. α-synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Genet. 2021, 17, e1009359.
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210.
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42.
- Fussi, N.; Höllerhage, M.; Chakroun, T.; Nykänen, N.P.; Rösler, T.W.; Koeglsperger, T.; Wurst, W.; Behrends, C.; Höglinger, G.U. Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell. Death Dis. 2018, 9, 757.
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119.
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192.
- Angot, E.; Steiner, J.A.; Lema Tomé, C.M.; Ekström, P.; Mattsson, B.; Björklund, A.; Brundin, P. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE 2012, 7, e39465.
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107.
- Orenstein, S.J.; Kuo, S.H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406.
- Ho, P.W.; Leung, C.T.; Liu, H.; Pang, S.Y.; Lam, C.S.; Xian, J.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 2020, 16, 347–370.
- Manzoni, C.; Mamais, A.; Roosen, D.A.; Dihanich, S.; Soutar, M.P.; Plun-Favreau, H.; Bandopadhyay, R.; Hardy, J.; Tooze, S.A.; Cookson, M.R.; et al. mTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin-1. Sci. Rep. 2016, 6, 35106.
- Obergasteiger, J.; Frapporti, G.; Lamonaca, G.; Pizzi, S.; Picard, A.; Lavdas, A.A.; Pischedda, F.; Piccoli, G.; Hilfiker, S.; Lobbestael, E.; et al. Kinase inhibition of G2019S-LRRK2 enhances autolysosome formation and function to reduce endogenous alpha-synuclein intracellular inclusions. Cell. Death Discov. 2020, 6, 45.
- Bravo-San Pedro, J.M.; Niso-Santano, M.; Gómez-Sánchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; López de Maturana, R.; Sanchez-Pernaute, R.; López de Munain, A.; et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell. Mol. Life Sci. 2013, 70, 121–136.
- Giaime, E.; Tong, Y.; Wagner, L.K.; Yuan, Y.; Huang, G.; Shen, J. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 2017, 96, 796–807.
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343.
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87.
- Geisler, S.; Holmström, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878.
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Höllerhage, M.; Höglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 865.
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342–378.
- Little, D.; Luft, C.; Mosaku, O.; Lorvellec, M.; Yao, Z.; Paillusson, S.; Kriston-Vizi, J.; Gandhi, S.; Abramov, A.Y.; Ketteler, R.; et al. A single cell high content assay detects mitochondrial dysfunction in iPSC-derived neurons with mutations in SNCA. Sci. Rep. 2018, 8, 9033.
- Park, J.H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 2020, 15, 5.
- Sampaio-Marques, B.; Felgueiras, C.; Silva, A.; Rodrigues, M.; Tenreiro, S.; Franssens, V.; Reichert, A.S.; Outeiro, T.F.; Winderickx, J.; Ludovico, P. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 2012, 8, 1494–1509.
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem. Cell. 2016, 19, 709–724.
- Bonello, F.; Hassoun, S.M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660.
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. Mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 2020, 16, 203–222.
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820.
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130.
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513.
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828.
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628.
- Ma, K.Y.; Fokkens, M.R.; Reggiori, F.; Mari, M.; Verbeek, D.S. Parkinson’s disease-associated VPS35 mutant reduces mitochondrial membrane potential and impairs PINK1/Parkin-mediated mitophagy. Transl. Neurodegener. 2021, 10, 19.
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594.
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits α-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front Aging Neurosci. 2017, 9, 308.
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752.
- Ivatt, R.M.; Sanchez-Martinez, A.; Godena, V.K.; Brown, S.; Ziviani, E.; Whitworth, A.J. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499.
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265.
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.; Tang, A.M.; Shaffra, F.; Li, Z.; et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330.
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394.
- Jinn, S.; Blauwendraat, C.; Toolan, D.; Gretzula, C.A.; Drolet, R.E.; Smith, S.; Nalls, M.A.; Marcus, J.; Singleton, A.B.; Stone, D.J. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum. Mol. Genet. 2019, 28, 3244–3254.
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 2017, 8, 15500.
- Grünewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Münchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, 1841–1843.
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743.
- Hunn, B.H.M.; Vingill, S.; Threlfell, S.; Alegre-Abarrategui, J.; Magdelyns, M.; Deltheil, T.; Bengoa-Vergniory, N.; Oliver, P.L.; Cioroch, M.; Doig, N.M.; et al. Impairment of Macroautophagy in Dopamine Neurons Has Opposing Effects on Parkinsonian Pathology and Behavior. Cell Rep. 2019, 29, 920–931.
- Rosenberg, R.N.; Lambracht-Washington, D.; Yu, G.; Xia, W. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016, 73, 867–874.
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199.
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342.
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98.
- Sanchez-Varo, R.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; De Castro, V.; Jimenez, S.; Ruano, D.; Vizuete, M.; et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 2012, 123, 53–70.
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158.
- Coffey, E.E.; Beckel, J.M.; Laties, A.M.; Mitchell, C.H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124.
- Moreau, K.; Fleming, A.; Imarisio, S.; Lopez Ramirez, A.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 2014, 5, 4998.
- Ando, K.; Tomimura, K.; Sazdovitch, V.; Suain, V.; Yilmaz, Z.; Authelet, M.; Ndjim, M.; Vergara, C.; Belkouch, M.; Potier, M.C.; et al. Level of PICALM, a key component of clathrin-mediated endocytosis, is correlated with levels of phosphotau and autophagy-related proteins and is associated with tau inclusions in AD, PSP and Pick disease. Neurobiol. Dis. 2016, 94, 32–43.
- Ando, K.; De Decker, R.; Vergara, C.; Yilmaz, Z.; Mansour, S.; Suain, V.; Sleegers, K.; de Fisenne, M.A.; Houben, S.; Potier, M.C.; et al. Picalm reduction exacerbates tau pathology in a murine tauopathy model. Acta Neuropathol. 2020, 139, 773–789.
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169.
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483.
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69.
- Tong, B.C.; Wu, A.J.; Huang, A.S.; Dong, R.; Malampati, S.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Zhu, Z.; Su, C.; et al. Lysosomal TPCN (two pore segment channel) inhibition ameliorates beta-amyloid pathology and mitigates memory impairment in Alzheimer disease. Autophagy 2021, 1–19.
- Zheng, X.; Lin, W.; Jiang, Y.; Lu, K.; Wei, W.; Huo, Q.; Cui, S.; Yang, X.; Li, M.; Xu, N.; et al. Electroacupuncture ameliorates beta-amyloid pathology and cognitive impairment in Alzheimer disease via a novel mechanism involving activation of TFEB (transcription factor EB). Autophagy 2021, 1–15.
- Chen, M.L.; Hong, C.G.; Yue, T.; Li, H.M.; Duan, R.; Hu, W.B.; Cao, J.; Wang, Z.X.; Chen, C.Y.; Hu, X.K.; et al. Inhibition of miR-331-3p and miR-9-5p ameliorates Alzheimer’s disease by enhancing autophagy. Theranostics 2021, 11, 2395–2409.
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69.
- Benito-Cuesta, I.; Ordóñez-Gutiérrez, L.; Wandosell, F. AMPK activation does not enhance autophagy in neurons in contrast to MTORC1 inhibition: Different impact on β-amyloid clearance. Autophagy 2021, 17, 656–671.
- Park, J.S.; Kim, D.H.; Yoon, S.Y. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016, 49, 337–342.
- Jiang, S.; Bhaskar, K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front Mol. Neurosci. 2020, 13, 586731.
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17, e12692.
- Silva, M.C.; Nandi, G.A.; Tentarelli, S.; Gurrell, I.K.; Jamier, T.; Lucente, D.; Dickerson, B.C.; Brown, D.G.; Brandon, N.J.; Haggarty, S.J. Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat. Commun. 2020, 11, 3258.
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714.
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front Aging Neurosci. 2019, 11, 311.
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412.
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951.
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806.
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38.
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368.
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
06 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No