 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ken H. Young | + 2840 word(s) | 2840 | 2021-11-23 10:09:03 |
Video Upload Options
Programmed death-ligand 1 (PD-L1) is one of the most classic immune checkpoint molecules. Cancer cells express PD-L1 to inhibit the activity of effector T cells’ cytotoxicity through programmed death 1 (PD-1) engagement in exposure to inflammatory cytokines. PD-L1 expression levels on cancer cells might affect the clinical response to anti-PD-1/PD-L1 therapies. Hence, understanding molecular mechanisms for regulating PD-L1 expression is essential for improving the clinical response rate and efficacy of PD-1/PD-L1 blockade. Posttranslational modifications (PTMs), including phosphorylation, glycosylation, ubiquitination, and acetylation, regulate PD-L1 stability, cellular translocation, and interaction with its receptor. A coordinated positive and negative regulation via PTMs is required to ensure the balance and function of the PD-L1 protein.
1. Introduction
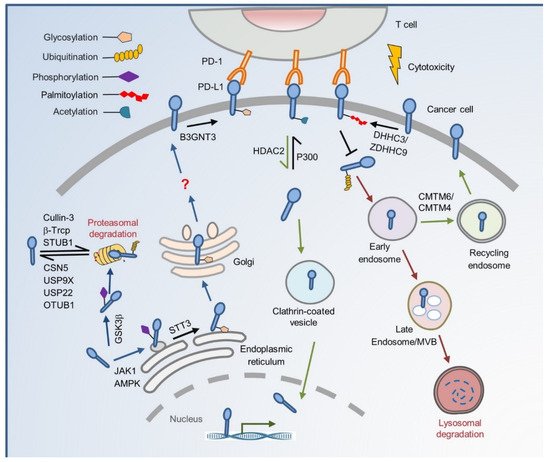
2. The Cradle: From De Novo Synthesis to Plasma Membrane
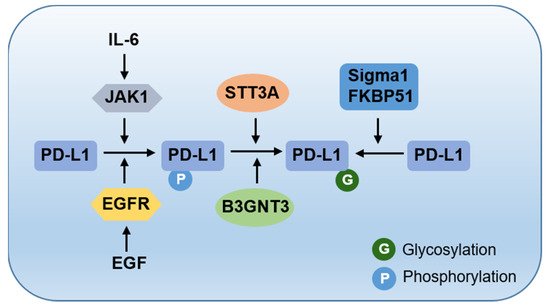
3. To the Grave: From Cell Membrane to Recycling and Lysosome Degradation
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668.
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034.
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319.
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813.
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833.
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356.
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452.
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4.
- Cha, J.-H.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Hung, M.-C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370.
- Zhang, J.; Dang, F.; Ren, J.; Wei, W. Biochemical Aspects of PD-L1 Regulation in Cancer Immunotherapy: (Trends in Biochemical Sciences 43, 1014-1032; 2018). Trends Biochem. Sci. 2019, 44, 557.
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672.
- Mowen, K.A.; David, M. Unconventional post-translational modifications in immunological signaling. Nat. Immunol. 2014, 15, 512–520.
- Horita, H.; Law, A.; Hong, S.; Middleton, K. Identifying Regulatory Posttranslational Modifications of PD-L1: A Focus on Monoubiquitinaton. Neoplasia 2017, 19, 346–353.
- Hsu, J.-M.; Li, C.-W.; Lai, Y.-J.; Hung, M.-C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018, 78, 6349–6353.
- Lippincott-Schwartz, J.; Roberts, T.H.; Hirschberg, K. Secretory Protein Trafficking and Organelle Dynamics in Living Cells. Annu. Rev. Cell Dev. Biol. 2000, 16, 557–589.
- Gomez-Navarro, N.; Miller, E. Protein sorting at the ER–Golgi interface. J. Cell Biol. 2016, 215, 769–778.
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957.
- Deng, S.; Zhou, X.; Xu, J. Checkpoints Under Traffic Control: From and to Organelles. Adv. Exp. Med. Biol. 2020, 1248, 431–453.
- Vagin, O.; Kraut, J.A.; Sachs, G. Role of N-glycosylation in trafficking of apical membrane proteins in epithelia. Am. J. Physiol. Physiol. 2009, 296, F459–F469.
- Moremen, K.W.; Tiemeyer, M.; Nairn, A. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462.
- Imperiali, B.; O’Connor, S.E. Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 1999, 3, 643–649.
- Breitling, J.; Aebi, M. N-Linked Protein Glycosylation in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013359.
- Varki, A. Evolutionary Forces Shaping the Golgi Glycosylation Machinery: Why Cell Surface Glycans Are Universal to Living Cells. Cold Spring Harb. Perspect. Biol. 2011, 3, a005462.
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632.
- Maher, C.M.; Thomas, J.D.; Haas, D.; Longen, C.G.; Oyer, H.M.; Tong, J.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2017, 16, 243–255.
- D’Arrigo, P.; Russo, M.; Rea, A.; Tufano, M.; Guadagno, E.; Del Basso De Caro, M.L.; Pacelli, R.; Hausch, F.; Staibano, S.; Ilardi, G.; et al. A regulatory role for the co-chaperone FKBP51s in PD-L1 expression in glioma. Oncotarget 2017, 8, 68291–68304.
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908.
- Chan, L.-C.; Li, C.-W.; Xia, W.; Hsu, J.-M.; Lee, H.-H.; Cha, J.-H.; Wang, H.-L.; Yang, W.-H.; Yen, E.-Y.; Chang, W.-C.; et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J. Clin. Investig. 2019, 129, 3324–3338.
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e607.
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nat. Cell Biol. 2018, 553, 91–95.
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and Cellular Roles of Ubiquitin-Specific Deubiquitinating Enzymes. Annu. Rev. Biochem. 2009, 78, 363–397.
- Lim, S.-O.; Li, C.-W.; Xia, W.; Cha, J.-H.; Chan, L.-C.; Wu, Y.; Chang, S.-S.; Lin, W.-C.; Hsu, J.-M.; Hsu, Y.-H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939.
- Jingjing, W.; Wenzheng, G.; Donghua, W.; Guangyu, H.; Aiping, Z.; Wenjuan, W. Deubiquitination and stabilization of programmed cell death ligand 1 by ubiquitin-specific peptidase 9, X-linked in oral squamous cell carcinoma. Cancer Med. 2018, 7, 4004–4011.
- Wang, Y.; Sun, Q.; Mu, N.; Sun, X.; Wang, Y.; Fan, S.; Su, L.; Liu, X. The deubiquitinase USP22 regulates PD-L1 degradation in human cancer cells. Cell Commun. Signal. 2020, 18, 112.
- Huang, X.; Zhang, Q.; Lou, Y.; Wang, J.; Zhao, X.; Wang, L.; Zhang, X.; Li, S.; Zhao, Y.; Chen, Q.; et al. USP22 Deubiquitinates CD274 to Suppress Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 1580–1590.
- Zhu, D.; Xu, R.; Huang, X.; Tang, Z.; Tian, Y.; Zhang, J.; Zheng, X. Deubiquitinating enzyme OTUB1 promotes cancer cell immunosuppression via preventing ER-associated degradation of immune checkpoint protein PD-L1. Cell Death Differ. 2020, 28, 1773–1789.
- Li, C.-W.; Lim, S.-O.; Chung, E.M.; Kim, Y.-S.; Park, A.H.; Yao, J.; Cha, J.-H.; Xia, W.; Chan, L.-C.; Kim, T.; et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33, 187–201.e10.
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386.
- Yang, Y.; Li, C.-W.; Chan, L.-C.; Wei, Y.; Hsu, J.-M.; Xia, W.; Cha, J.-H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864.
- Daassi, D.; Mahoney, K.M.; Freeman, G.J. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215.
- Poggio, M.; Hu, T.; Pai, C.-C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 2019, 177, 414–427.e13.
- Moroishi, T.; Hayashi, T.; Pan, W.-W.; Fujita, Y.; Holt, M.V.; Qin, J.; Carson, D.A.; Guan, K.-L. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 2016, 167, 1525–1539.e17.
- Castello, A.; Rossi, S.; Toschi, L.; Mansi, L.; Lopci, E. Soluble PD-L1 in NSCLC Patients Treated with Checkpoint Inhibitors and Its Correlation with Metabolic Parameters. Cancers 2020, 12, 1373.
- Frigola, X.; Inman, B.A.; Lohse, C.M.; Krco, C.J.; Cheville, J.C.; Thompson, R.H.; Leibovich, B.; Blute, M.L.; Dong, H.; Kwon, E.D. Identification of a Soluble Form of B7-H1 That Retains Immunosuppressive Activity and Is Associated with Aggressive Renal Cell Carcinoma. Clin. Cancer Res. 2011, 17, 1915–1923.
- Chen, Y.; Wang, Q.; Shi, B.; Xu, P.; Hu, Z.; Bai, L.; Zhang, X. Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine 2011, 56, 231–238.
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 480–492.
- Okuma, Y.; Hosomi, Y.; Nakahara, Y.; Watanabe, K.; Sagawa, Y.; Homma, S. High plasma levels of soluble programmed cell death ligand 1 are prognostic for reduced survival in advanced lung cancer. Lung Cancer 2017, 104, 1–6.
- Kruger, S.; Legenstein, M.-L.; Rösgen, V.; Haas, M.; Modest, D.P.; Westphalen, C.B.; Ormanns, S.; Kirchner, T.; Heinemann, V.; Holdenrieder, S.; et al. Serum levels of soluble programmed death protein 1 (sPD-1) and soluble programmed death ligand 1 (sPD-L1) in advanced pancreatic cancer. OncoImmunology 2017, 6, e1310358.
- Gong, B.; Kiyotani, K.; Sakata, S.; Nagano, S.; Kumehara, S.; Baba, S.; Besse, B.; Yanagitani, N.; Friboulet, L.; Nishio, M.; et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non–small cell lung cancer. J. Exp. Med. 2019, 216, 982–1000.
- Marsh, M.; McMahon, H.T. The Structural Era of Endocytosis. Science 1999, 285, 215–220.
- Aguilar, R.C.; Wendland, B. Endocytosis of membrane receptors: Two pathways are better than one. Proc. Natl. Acad. Sci. USA 2005, 102, 2679–2680.
- Burr, M.L.; Sparbier, C.E.; Chan, Y.C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 2017, 549, 101–105.
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; De Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110.
- Linder, M.E.; Deschenes, R. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84.
- Yang, Y.; Hsu, J.-M.; Sun, L.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Wei, Y.; Xia, W.; Hou, J.; Qiu, Y.; et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019, 29, 83–86.
- Yao, H.; Lan, J.; Li, C.; Shi, H.; Brosseau, J.-P.; Wang, H.; Lu, H.; Fang, C.; Zhang, Y.; Liang, L.; et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 2019, 3, 306–317.
- Parra, E.R.; Villalobos, P.; Rodriguez-Canales, J. The Multiple Faces of Programmed Cell Death Ligand 1 Expression in Malignant and Nonmalignant Cells. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 287–294.
- Satelli, A.; Batth, I.S.; Brownlee, Z.; Rojas, C.; Meng, Q.H.; Kopetz, S.; Li, S. Potential role of nuclear PD-L1 expression in cell-surface vimentin positive circulating tumor cells as a prognostic marker in cancer patients. Sci. Rep. 2016, 6, 28910.
- Schulz, D.; Streller, M.; Piendl, G.; Brockhoff, G.; E Reichert, T.; Menevse, A.N.; Beckhove, P.; Hautmann, M.G.; Bauer, R.J.; Ettl, T. Differential localization of PD-L1 and Akt-1 involvement in radioresistant and radiosensitive cell lines of head and neck squamous cell carcinoma. Carcinogenesis 2020, 41, 984–992.
- Ghebeh, H.; Lehe, C.; Barhoush, E.; Al-Romaih, K.; Tulbah, A.; Al-Alwan, M.; Hendrayani, S.-F.; Manogaran, P.; Alaiya, A.; Al-Tweigeri, T.; et al. Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breast cancer cells: Role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. 2010, 12, R48.
- Du, W.; Zhu, J.; Zeng, Y.; Liu, T.; Zhang, Y.; Cai, T.; Fu, Y.; Zhang, W.; Zhang, R.; Liu, Z.; et al. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Differ. 2020, 28, 1284–1300.
- Gao, Y.; Nihira, N.T.; Bu, X.; Chu, C.; Zhang, J.; Kolodziejczyk, A.; Fan, Y.; Chan, N.T.; Ma, L.; Liu, J.; et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol. 2020, 22, 1064–1075.
- Wang, H.; Yao, H.; Li, C.; Shi, H.; Lan, J.; Li, Z.; Zhang, Y.; Liang, L.; Fang, J.-Y.; Xu, J. HIP1R targets PD-L1 to lysosomal degradation to alter T cell–mediated cytotoxicity. Nat. Chem. Biol. 2019, 15, 42–50.


