 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emre Bektik | + 3416 word(s) | 3416 | 2020-08-10 04:16:17 | | | |
| 2 | Bruce Ren | Meta information modification | 3416 | 2020-08-24 11:32:06 | | | | |
| 3 | Bruce Ren | Meta information modification | 3416 | 2020-10-27 10:03:54 | | |
Video Upload Options
Atrial fibrillation (AF) is a type of sustained arrhythmia in humans often characterized by devastating alterations to the cardiac conduction system as well as the structure of the atria. AF can lead to decreased cardiac function, heart failure, and other complications. Long non-coding RNAs (lncRNAs) have been shown to play important roles in the cardiovascular system, including AF; however, a large group of lncRNAs is not conserved between mouse and human. Furthermore, AF has complex networks showing variations in mechanisms in different species, making it challenging to utilize conventional animal models to investigate the functional roles and potential therapeutic benefits of lncRNAs for AF. Fortunately, pluripotent stem cell (PSC)-derived cardiomyocytes (CMs) offer a reliable platform to study lncRNA functions in AF because of certain electrophysiological and molecular similarities with native human CMs.
1. Introduction
Atrial fibrillation (AF) is the most common form of cardiac arrhythmias among heart patients [1], which ultimately leads to heart failure and other complications, which increase morbidity and mortality [2]. The pathology of AF is characterized by remodeling of the atrial electrical
system—often mediated by abnormalities in ion channels [3][4][5]. One major feature of atrial remodeling in AF is the shortened refractory period leading to reentry due to reduced action potential duration (APD) [6][7]. The cause of AF is not fully understood, as genetic background, lifestyle, hypertension, or other underlying pathological conditions can all play a role [8].
With the advancement of new RNA sequencing technologies, a large set of noncoding RNAs (ncRNAs) were characterized in AF patients, which were believed to have essential roles in both atrial development and disease [9]. NcRNAs are non-protein coding RNAs that are divided into two major groups: small ncRNAs less than 200 nt in length and long ncRNAs (lncRNAs) more than 200 nt in length. MicroRNAs (miRNAs), a subgroup of short non-coding RNAs, have been widely studied in AF and shown to play an essential role in this condition [10][11]. In past decade, the identification of lncRNAs opened a new avenue of investigation in AF research. Accumulating evidence shows that lncRNAs play key roles in several cardiovascular diseases, including AF [12]. Consequently, it is important to determine how they function in AF development and whether they could be used to treat AF. Indeed, recent studies have shown that lncRNAs can serve as molecular biomarkers in AF patients and possess the potential to be therapeutic targets [13][14]. Therefore, the identification and characterization of new lncRNAs could shed light on the mechanisms behind AF and may serve to prevent, diagnose, and, ultimately, treat AF. Because the causes of AF are highly variable between human and animal models, it is essential to develop appropriate disease models using human cells or tissues [15]. Human pluripotent stem cells (hPSCs) are emerging as an essential system to model AF, whether they are derived from patients or genetically manipulated in vitro.
2. LncRNAs in Atrial Remodeling and Development of AF
Although lncRNAs have been studied and shown to function in various heart diseases, their function in the pathogenesis of AF is not well-understood. AF development is a complicated process, yet it is primarily caused by either structural remodeling or electrical remodeling of the atria, which disrupts the contraction of atrial cardiomyocytes [16]. Table 1 summarizes known lncRNAs that participate in atrial remodeling and AF development.
Table 1. LncRNAs in the development and pathophysiological remodeling of AF.
|
Development of AF |
|||
|
LncRNA |
Expression in AF |
Mechanism of Action |
Study Model |
|
RP11-99E15.2 [17] |
ND |
May be involved in AF by regulating extracellular matrix binding via interactions with ITGB3 |
Patient (AF) |
|
RP3-523K23.2 [17] |
ND |
May be involved in AF by regulating transcription of HSF2 |
Patient (AF) |
|
AK055347 [18] |
Up |
Dysregulating mitochondrial energy production by regulating mitochondrial Cyp450, ATP synthase, and MSS51 |
Patient (AF) |
|
Structural Remodeling |
|||
|
Up |
Regulating miR-128-3p/Sp1/TGF-b1/Smad axis by sponging miR-128-3p |
Patient (AF) or Atrial fibroblast; Mouse heart (Ang-II) |
|
|
GAS5 [21] |
Down |
Inhibiting ALK5 and suppresses cell proliferation |
Patient (AF) or AC16 |
|
PCAT1 [22] |
Up |
Promoting fibroblast proliferation through targeting TGF-b1 |
Patient (AF) or AC16 |
|
MIAT [23] |
Up |
Alleviating AF and reducing atrial fibrosis by suppressing miR-133-3p |
Patient (AF); Rat (electrical stimulation) |
|
NRON [24] |
Up |
Inhibiting NFAT localization to nucleus, thus suppresses IL-12 and macrophage switch from M2 to M1. |
Mouse atrial CM (AngII) |
|
TCONS_00032546 [25] |
Down |
Related to RAS-mediated neuronal remodeling in cardiac fat pads |
Canine heart (atrial tachypacing) |
|
TCONS_00026102 [25] |
Down |
Related to RAS-mediated neuronal remodeling in cardiac fat pads |
Canine heart (atrial tachypacing) |
|
Electrical Remodeling |
|||
|
PANCR [26] |
ND |
Regulating PITX2, an AF-related gene, but not studied in AF directly |
Human ESC-CM |
|
TCONS_00075467 [27] |
Down |
Upregulating of it results with increased sponging of miR328, thus increasing CACNA1C levels |
Rabbit right atria (AF) |
|
KCNQ1OT1 [28] |
Up |
Downregulating of it results with decreased sponging of miR384, thus decreasing CACNA1C levels |
Mouse heart (AngII) or CM |
|
NPPA-AS1 [17] |
Up |
Modulating cardiac contraction genes (e.g., NPPA, PLCE1, TNNC1, TNN1). |
Patient (AF) |
|
lncRNA-HBL1 [29] |
Up |
Downregulating miR-1, an AF-related gene, but not studied in AF directly |
Human iPSC-CM |
2.1. LncRNAs in the Development of AF
A number of reports have described differentially expressed lncRNAs in AF [30][31]. Mei et al. [31]demonstrated that 182 lncRNAs were differentially expressed in left atrial tissue samples from patients with AF compared to patients with normal sinus rhythm. Ruan et al. [32] performed microarray analyses on AF patient samples to examine the expression profile of lncRNAs and found 219 differentially expressed lncRNAs compared to AF-free patients. They postulated that the lncRNAs identified in their study were responsible for AF initiation by promoting electrical remodeling and altering the renin-angiotensin system (RAS). In another study, Xu et al. showed 177 differentially expressed lncRNAs in AF patients. They also analyzed co-expression profiles of lncRNAs with mRNAs and found that the transcriptional regulators GATA1, TAF7, and EBF1 were involved in expression of AF-related lncRNAs. Indeed, previous findings have shown that GATA1 and TAF1 have known roles in AF pathology [33][34]. Ke et al. also performed a differential expression analysis of lncRNAs in the left and right atrium of AF patients and identified two AF-linked lncRNAs (RP11-99E15.2 and RP3-523K23.2) that regulate heat shock factor 2 (HSF2), which is an important player in hypertension-induced HF. Furthermore, Cheng et al. examined lncRNA expression profiles in the left atrial appendage and left atrial tissue surrounding the pulmonary veins, where AF is believed to be initiated, and identified 94 differentially expressed lncRNAs. Among them, AK055347 was found to be the most significantly altered. Knockdown of AK055347 inhibited expression of the mitochondrial genes Cyp450, ATP synthase, and MASS51, resulting in decreased viability of H9C2 cardiomyocytes. These findings suggested that AK055347 may be a regulator of AF by affecting mitochondrial energy production.
In summary, lncRNAs have been identified as being involved in AF by establishing a connection between their expression and that of AF-related gene networks. Therefore, we will now focus on lncRNAs associated with structural and electrical remodeling in AF.
2.2. LncRNAs in Atrial Structural Remodeling
Fibrosis is a hallmark of structural remodeling of the atria and is a multifactorial process. LncRNAs were identified to interact with key factors associated with the development of atrial fibrosis and AF such as TGF-b1, which is the most commonly cited factor involved in fibrosis. Zhao et al. found 57 differentially expressed lncRNAs in epicardial adipose tissue from AF patients compared to patients with normal sinus rhythm. Compelling evidence showed that lncRNAs secreted from epicardial adipose tissue diffuse into the myocardium and induce atrial fibrosis . Another study showed that AF patients had increased amounts of adipose tissue on their myocardium and increased levels of the fibrotic markers TGF-b1 and Smad2 in this tissue [33]. Zhao et al. identified multiple lncRNAs connected with protein-coding genes that are known to play role in atrial fibrosis. One of these lncRNAs was plasmacytoma variant translocation 1 (PVT1), whose expression was found to associate with genes related to lipid metabolism, inflammation, and TGF-b1-induced epithelial-to-mesenchymal (EMT) transition such as NOS3, TTC3, PDLIM1, and SP1 . In particular, PVT1 expression was elevated in atrial muscle tissue from AF patients . Overexpression of PVT1 induced fibrosis by acting as a sponge for miR-128-3p; thereby, regulating the miR-128-3p/Sp1/TGF-b1/Smad axis, leading to SP1-mediated activation of the TGF-b1/Smad pathway and increased production of collagen I and II. Conversely, inhibition of PVT1 reversed fibrosis.
ALK5, a downstream target of TGF-b1, is known to regulate the proliferation of cells [34], including cardiac fibroblasts. Growth inhibitory specificity (GAS5) is a lncRNA that inhibits ALK5 in cardiomyocytes . GAS5 expression was decreased in atrial appendage samples from AF patients. Knockdown of GAS5 increased cell growth, while its overexpression inhibited growth of AC16 cells in vitro. Prostate cancer associated transcript-1 (PCAT1) is another lncRNA that has been identified during the fibrotic remodeling of AF. PCAT1 expression levels were higher in atrial appendage samples from AF patients. PCAT1 promoted fibroblast proliferation by targeting TGF-b1. Aside from its function in cardiac hypertrophy and fibrosis, MIAT has recently been shown to play a role in AF through repression of miR-133-3p . MIAT expression was elevated in the atrial tissues from a rat model of AF, while miR-133-3p expression was decreased. Knockdown of MIAT alleviated AF and reduced the duration of fibrillation episodes as well as promoting increased atrial function and suppressing cardiomyocyte apoptosis. On the other hand, MIAT knockdown was shown to suppress AF-induced atrial fibrosis.
Macrophages are also involved in atrial fibrosis. M1 macrophages arrive to the site of injury and clear the cellular debris, while M2 macrophages help regulate the tissue-healing process. It was shown that a switch from M1 to M2 macrophage types prevents cardiac remodeling and improves heart function . Noncoding repressor of NFAT (NRON) is a lncRNA that is normally recruited to the promoter of interleukin-12 (IL-12), which is known to induce the switch from M2 to M1 types, leading to atrial fibrosis . NRON was found to inhibit nuclear localization of NFAT, thus inhibiting IL-12, which reversed macrophage switching and decreased atrial fibrosis. Another study that examined lncRNAs associated with immune signaling, identified co-expression networks between up-regulated mRNAs and lncRNAs in lymphocytes collected from AF and non-AF patients [35]. Those networks were related to tumor necrosis factor (TNF), toll-like receptor (TLR), and NF-κβ signaling pathways. In short, lncRNAs from lymphocytes were linked with cellular processes including collagen synthesis, oxidative stress, inflammation, and apoptosis, which are all involved in the development of atrial fibrosis.
In addition to the above, increased sympathetic neuronal activity has been recorded in patients prior to the development of post-operative AF [36]. Interestingly, RAS has been shown to interact with the autonomic nervous system and is involved in neuronal remodeling. Wang et al. analyzed lncRNAs in cardiac fat pads of canines with or without AF and found that aberrantly expressed lncRNAs were related to neuronal development, differentiation, and degeneration. Among the identified lncRNAs, they showed that in vivo inhibition of two lncRNAs (TCONS_00032546 and TCONS_00026102) related to neuronal remodeling, either shortened or prolonged the atrial refractory period, resulting in the increased occurrence or prevention of AF. In addition, the expression of these lncRNAs was negatively correlated with CCND1, FGF19, FGF4, FGF3, and SLC25A4 expression as well as the genes neighboring these lncRNAs. Together, these studies suggest that lncRNAs may contribute to the development of AF through RAS-mediated neuronal remodeling.
2.3. LncRNAs in Atrial Electrical Remodeling
LncRNAs are also involved in electrical remodeling in AF, although this connection is less well-established than their involvement with structural remodeling. The major drivers of electrical remodeling are shortening of the atrial refractory period and action potential duration. One of the genes identified in AF is PITX2, which is also associated with heart development. Holmes et al. found that mice predisposed to developing AF had relatively low levels of PITX2. PITX2 affected cardiac ion channels to alter the atrial refractory period. In addition, PANCR, an upstream lncRNA targeting PITX2, was identified . However, PANCR has not been directly linked with development of AF; yet, because of the role of PITX2 in AF, PANCR can be considered an AF-related lncRNA .
Li et al. examined lncRNAs in AF and non-AF rabbits and found that silencing of the lncRNA TCONS_00075467 resulted in a shortened atrial refractory period and action potential duration. This effect was likely due to its role as a sponge for miR-328, which silences its inhibitory function on target mRNAs. Because of the lack of this lncRNA, miR-328 expression increases, resulting in downregulation of CACNA1C, an L-type calcium ion channel. Indeed, dysregulation of CACNA1C has been previously shown to be involved in the development of AF through regulation of RAS [37][38]. Activation of RAS has been shown to increase left atrial pressure via AngII in hypertension and heart failure. Moreover, prolongation of RAS activation induces high levels of angiotensin-converting enzyme (ACE) and AngII receptors, resulting in inflammation and fibrosis or structural remodeling [39][40].
In mice with AngII-induced AF, Shen et al. identified an overexpressed lncRNA, KCNQ1 overlapping transcript 1 (KCNQ1OT1). KCNQ1OT1 acts as a sponge to miR-384, which targets and silences CACNA1C. Overexpression of KCNQ1OT1 inhibits the silencing effect of miR-384, leading to elevated CACNA1C levels and the development of AF.
Ke et al. analyzed RNAseq data in left and right atrial appendages from patients with or without AF and identified key RNAs linked with AF. They predicted lncRNAs that potentially regulate adjacent protein-coding genes and found that lncRNA NPPA-AS1, RP11-99E15.2, and RP3-523K23.2 could interact with NPPA, ITGB3, and HSF2, respectively, and may be involved in the pathogenesis of AF. Particularly, NPPA-AS1 was found to be co-expressed with six contractile genes, including NPPA, PLCE1, TACR1, GSTO1, TNNC1, and TNN1, suggesting that NPPA-AS1 contributes to AF pathogenesis through the modulation of cardiac contraction.
3. Deciphering LncRNA Function in Atrial Fibrillation by hPSC Disease Modeling
A large-scale evolutionary study showed that many lncRNAs have very limited sequential conservation unlike protein-coding counterparts [41][42][43][44]. Additionally, multidisciplinary analyses of lncRNAs from different species showed the vast majority of lncRNAs show minimal conservation [45]. Consequently, this limited conservation between species restricts the effective study of many lncRNAs in human AF. Also, the lack of a mechanistic understanding of AF development is an obstacle to understanding AF substrates. The most valid method is to obtain tissue samples from patients to study lncRNAs; however, there is a limited supply of these tissues from patients, let alone healthy individuals [46][47][48]. Therefore, the study of lncRNAs in AF are increasingly dependent on human pluripotent stem cell (hPSC)-derived atrial cardiomyocytes, which allows both large-scale experiments and genetic manipulation (Figure 1).
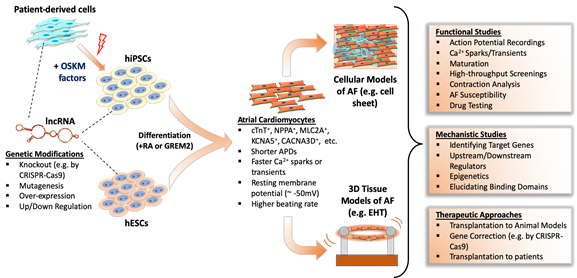
Figure 1. Studying function and mechanism of lncRNAs in human pluripotent stem cell-based atrial fibrillation models.
3.1. Differentiation and Characterization of hPSC-Derived Atrial Cardiomyocytes
Recent advances in atrial lineage differentiation of pluripotent stem cells has allowed the culture of pure and functional atrial myocytes to model AF [49][42][43][44][50]. Zheng et al. showed that exogenous activation of retinoic acid (RA) signaling in differentiating human embryonic stem cells (hESCs) promoted an atrial phenotype as assessed by action potential characteristics and Ca2+ handling. Conversely, inhibition of RA signaling promoted a more ventricular differentiation program. Later, Cyganek et al. further examined the molecular, cellular, and functional properties of hiPSC-derived atrial and ventricular cardiomyocytes by manipulating RA signaling in plated cells and in engineered heart muscle (EHM). By studying cell structure, action potentials, calcium fluctuations, and the transcriptome/proteome as well as contractile characteristics of EHM, they showed that the RA-dependent differentiation of hiPSC-CMs produced functionally relevant cells to model human atrial diseases. Alternatively, atrial differentiation of hESCs by a bone morphogenic protein antagonist, GREMLIN 2 (GREM 2), also caused atrial differentiation of mouse ESCs [51]; however, this method has not been validated in human ESCs despite the role of GREM2 in heart development being conserved between species [52]. Overall, these findings suggest that hPSC-derived atrial cardiomyocytes are suitable for atrial disease modeling and drug screening.
3.2. Disease Modeling of AF Using hPSCs
The development of atrial differentiation methods has allowed modeling of, and drug screening for, AF. For example, Laksman et al. [53] generated an AF model from hESC-derived atrial cardiomyocytes by RA activation during mesodermal differentiation. They performed optical mapping on atrial cell sheets, induced AF by rapid pacing protocol or burst pacing, and successfully showed AP propagation and reentry patterns similar to those observed in AF. Importantly, their drug testing with flecainide and dofetilide modulated reentrant arrhythmic rotor activation toward a non-AF phenotype, underscoring the reliability of these drugs in AF treatment. In a recent study by Benzoni et al. [54], a familial form of AF was modeled using patient-derived iPSC-CMs. They generated several hiPSC cell lines from patients with a persistent, untreatable AF that were not responsive to anti-arrhythmic drugs. To identify mutations in those patients, they performed whole exome sequencing and found more than 100 variations between three AF patients, of which only a few were related to genes previously linked with AF (ZFHX3) or the heart (PDE4DIP, CNN2, RYR3, NEFM, FLNC, and MYLK). Because of the complexity of AF in these patients, they decided to investigate the molecular mechanisms of AF in hiPSC-derived atrial cardiomyocytes. Functional characterization by electrophysiology analyses revealed higher beating rates due to the increased contribution of hyperpolarization activated pacemaker current (If) and L-type Ca2+ channel current (ICaL) in patient-derived cardiomyocytes compared to healthy, control cells. Also, patient iPSC-CMs showed a significantly prolonged APD, and under cardiac stress, larger amplitudes of delayed after-depolarization (DAD) with more frequent ectopic beats than in control iPSC-CMs.
Hong et al. [55] also developed an AF model using patient-derived iPSCs to characterize the electrophysiological properties of a known familial AF linked with an E428K mutation on SCN5A (sodium voltage-gated alpha subunit 5). Atrial iPSC-CMs with this mutation showed an increased window for the late sodium current (INa,L), increased beating rate, prolonged APD, and spontaneous arrhythmogenic activity. Interestingly, ranolazine treatment reversed the abnormal phenotypes in mutant atrial iPSC-CMs.
The above examples prove that hPSC-derived cardiomyocytes might be a helpful resource to study the function of coding or non-coding genes in AF. Although differential gene expression analyses in human atrial tissue from AF patients identified potential lncRNAs involved in AF, those studies did not fully elucidate the role of lncRNAs in the development of this disease and did not determine the molecular mechanisms underlying AF. Additionally, the limited conservation of lncRNAs between species and the variability of atrial electrophysiology and cardiac anatomy have not allowed comprehensive studies of many lncRNAs in AF, thus, limiting the translational potential of animal models. Therefore, hPSCs may offer a reliable resource to identify early mechanisms and substrates of AF, in particular, those controlled by lncRNAs.
3.3. Use of hPSCs for the Study of LncRNAs in AF
To date, there is a lack of reports describing the use of hPSC-CMs to study the role of lncRNAs in the development of AF; however, there are some studies that employ iPSCs to identify the molecular mechanisms of lncRNAs, which are either directly or indirectly related to AF pathogenesis.
Heart Brake LncRNA 1 (lncRNA-HBL1) was initially described in human iPSCs by Liu et al. [135] in cardiomyocyte development. LncRNA-HBL1 overexpression suppressed cardiac differentiation of iPSCs by sequestering hsa-miR-1, an essential cardiac enriched miRNA. Although this study was not directly linked to AF, previous findings showed the importance of miR-1 in AF development. miR-1 levels were reduced in the left atrium of AF patients, resulting in increased inward rectifier potassium channel (Kir2.1) [56], and miR-1 was found to accelerate shortening of the atrial effective refractory period (AERP) in a rabbit model, resulting in increased AF susceptibility by down-regulation of KCNE1 and KCNB2 [57]. These findings suggest that lncRNA-HBL1 might have a role in AF development via regulation of miR1.
NPPA-AS1 has been shown to play a role in AF via modulation of atrial contractile genes . Celik et al. [58] studied the molecular mechanism of this lncRNA in iPSC-derived cardiomyocytes (iPSC-CMs), which contained a mixture of atrial, ventricular, and nodal CMs. They found that NPPA-AS1 was localized to the nucleus in iPSC-CMs. They also showed that NPPA-AS1, which is highly enriched in atrial tissue, negatively regulated NPPA expression by enhancing binding of the RE1-silencing transcription factor (REST) to the NPPA promoter, resulting in suppression of NPPA gene expression. NPPA-AS1 expression levels were relatively lower compared to NPPA levels in human atrial heart tissue, suggesting that its mode of action is limited to promoter activation, but not dimer formation with NPPA mRNAs. To evaluate the therapeutic potential of NPPA-AS1 inhibition, they tested antisense oligonucleotides (GapmeRs) in mouse hearts and showed that NPPA-AS1 silencing increased expression levels of Nppa. These observations suggest that NPPA-AS1 might be a therapeutic target to regulate NPPA expression in various heart disease conditions, including AF.
References
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. Heart J. 2016, 37, 2893–2962, doi:10.1093/eurheartj/ehw210.
- Conen, D.; Chae, C.U.; Glynn, R.J.; Tedrow, U.B.; Everett, B.M.; Buring, J.E.; Albert, C.M. Risk of death and cardiovascular events in initially healthy women with new-onset atrial fibrillation. JAMA 2011, 305, 2080–2087, doi:10.1001/jama.2011.659.
- Santulli, G.; D’Ascia, S.L.; D’Ascia, C. Development of atrial fibrillation in recipients of cardiac resynchronization therapy: The role of atrial reverse remodelling. Can. J. Cardiol. 2012, 28, 245, doi:10.1016/j.cjca.2011.11.001.
- Brundel, B.J.; Van Gelder, I.C.; Henning, R.H.; Tuinenburg, A.E.; Wietses, M.; Grandjean, J.G.; Wilde, A.A.; Van Gilst, W.H.; Crijns, H.J. Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: Differential regulation of protein and mRNA levels for K+ channels. J. Am. Coll. Cardiol. 2001, 37, 926–932, doi:10.1016/s0735-1097(00)01195-5.
- Xie, W.; Santulli, G.; Guo, X.; Gao, M.; Chen, B.X.; Marks, A.R. Imaging atrial arrhythmic intracellular calcium in intact heart. J. Mol. Cell. Cardiol. 2013, 64, 120–123, doi:10.1016/j.yjmcc.2013.09.003.
- Kapur, S.; Macrae, C.A. The developmental basis of adult arrhythmia: Atrial fibrillation as a paradigm. Front. Physiol. 2013, 4, 221, doi:10.3389/fphys.2013.00221.
- D’Ascia, S.L.; D’Ascia, C.; Marino, V.; Lombardi, A.; Santulli, R.; Chiariello, M.; Santulli, G. Cardiac resynchronisation therapy response predicts occurrence of atrial fibrillation in non-ischaemic dilated cardiomyopathy. Int. J. Clin. Pract. 2011, 65, 1149–1155, doi:10.1111/j.1742-1241.2011.02732.x.
- Shi, K.H.; Tao, H.; Yang, J.J.; Wu, J.X.; Xu, S.S.; Zhan, H.Y. Role of microRNAs in atrial fibrillation: New insights and perspectives. Cell. Signal. 2013, 25, 2079–2084, doi:10.1016/j.cellsig.2013.06.009.
- Babapoor-Farrokhran, S.; Gill, D.; Rasekhi, R.T. The role of long noncoding RNAs in atrial fibrillation. Heart Rhythm. 2020, 17, 1043–1049, doi:10.1016/j.hrthm.2020.01.015.
- Dawson, K.; Wakili, R.; Ordog, B.; Clauss, S.; Chen, Y.; Iwasaki, Y.; Voigt, N.; Qi, X.Y.; Sinner, M.F.; Dobrev, D.; et al. MicroRNA29: A mechanistic contributor and potential biomarker in atrial fibrillation. Circulation 2013, 127, 1466–1475, doi:10.1161/CIRCULATIONAHA.112.001207.
- McManus, D.D.; Lin, H.; Tanriverdi, K.; Quercio, M.; Yin, X.; Larson, M.G.; Ellinor, P.T.; Levy, D.; Freedman, J.E.; Benjamin, E.J. Relations between circulating microRNAs and atrial fibrillation: Data from the Framingham Offspring Study. Heart Rhythm. 2014, 11, 663–669, doi:10.1016/j.hrthm.2014.01.018.
- Su, Y.; Li, L.; Zhao, S.; Yue, Y.; Yang, S. The long noncoding RNA expression profiles of paroxysmal atrial fibrillation identified by microarray analysis. Gene 2018, 642, 125–134, doi:10.1016/j.gene.2017.11.025.
- Xu, Y.; Huang, R.; Gu, J.; Jiang, W. Identification of long non-coding RNAs as novel biomarker and potential therapeutic target for atrial fibrillation in old adults. Oncotarget 2016, 7, 10803–10811, doi:10.18632/oncotarget.7514.
- Hobuss, L.; Bar, C.; Thum, T. Long Non-coding RNAs: At the Heart of Cardiac Dysfunction? Front. Physiol. 2019, 10, 30, doi:10.3389/fphys.2019.00030.
- Tucker, N.R.; Clauss, S.; Ellinor, P.T. Common variation in atrial fibrillation: Navigating the path from genetic association to mechanism. Cardiovasc. Res. 2016, 109, 493–501, doi:10.1093/cvr/cvv283.
- Nattel, S.; Harada, M. Atrial remodeling and atrial fibrillation: Recent advances and translational perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345, doi:10.1016/j.jacc.2014.02.555.
- Ke, Z.P.; Xu, Y.J.; Wang, Z.S.; Sun, J. RNA sequencing profiling reveals key mRNAs and long noncoding RNAs in atrial fibrillation. J. Cell. Biochem. 2019, doi:10.1002/jcb.29504.
- Chen, G.; Guo, H.; Song, Y.; Chang, H.; Wang, S.; Zhang, M.; Liu, C. Long noncoding RNA AK055347 is upregulated in patients with atrial fibrillation and regulates mitochondrial energy production in myocardiocytes. Mol. Med. Rep. 2016, 14, 5311–5317, doi:10.3892/mmr.2016.5893.
- Zhao, L.; Ma, Z.; Guo, Z.; Zheng, M.; Li, K.; Yang, X. Analysis of long non-coding RNA and mRNA profiles in epicardial adipose tissue of patients with atrial fibrillation. Biomed. Pharmacother. 2020, 121, 109634, doi:10.1016/j.biopha.2019.109634.
- Cao, F.; Li, Z.; Ding, W.M.; Yan, L.; Zhao, Q.Y. LncRNA PVT1 regulates atrial fibrosis via miR-128-3p-SP1-TGF-beta1-Smad axis in atrial fibrillation. Mol. Med. 2019, 25, 7, doi:10.1186/s10020-019-0074-5.
- Lu, J.; Xu, F.Q.; Guo, J.J.; Lin, P.L.; Meng, Z.; Hu, L.G.; Li, J.; Li, D.; Lu, X.H.; An, Y. Long noncoding RNA GAS5 attenuates cardiac fibroblast proliferation in atrial fibrillation via repressing ALK5. Eur. Rev. Med. Pharmacol Sci. 2019, 23, 7605–7610, doi:10.26355/eurrev_201909_18883.
- Chen, Q.; Feng, C.; Liu, Y.; Li, Q.F.; Qiu, F.Y.; Wang, M.H.; Shen, Z.D.; Fu, G.S. Long non-coding RNA PCAT-1 promotes cardiac fibroblast proliferation via upregulating TGF-beta1. Eur. Rev. Med. Pharmacol Sci. 2019, 23, 10517–10522, doi:10.26355/eurrev_201912_19692.
- Yao, L.; Zhou, B.; You, L.; Hu, H.; Xie, R. LncRNA MIAT/miR-133a-3p axis regulates atrial fibrillation and atrial fibrillation-induced myocardial fibrosis. Mol. Biol. Rep. 2020, 47, 2605–2617, doi:10.1007/s11033-020-05347-0.
- Sun, F.; Guo, Z.; Zhang, C.; Che, H.; Gong, W.; Shen, Z.; Shi, Y.; Ge, S. LncRNA NRON alleviates atrial fibrosis through suppression of M1 macrophages activated by atrial myocytes. Biosci. Rep. 2019, 39, doi:10.1042/BSR20192215.
- Wang, W.; Wang, X.; Zhang, Y.; Li, Z.; Xie, X.; Wang, J.; Gao, M.; Zhang, S.; Hou, Y. Transcriptome analysis of canine cardiac fat pads: Involvement of two novel long non-coding RNAs in atrial fibrillation neural remodeling. J. Cell. Biochem. 2015, 116, 809–821, doi:10.1002/jcb.25037.
- Holmes, A.P.; Kirchhof, P. Pitx2 Adjacent Noncoding RNA: A New, Long, Noncoding Kid on the 4q25 Block. Circ. Arrhythm. Electrophysiol. 2016, 9, e003808, doi:10.1161/CIRCEP.115.003808.
- Li, Z.; Wang, X.; Wang, W.; Du, J.; Wei, J.; Zhang, Y.; Wang, J.; Hou, Y. Altered long non-coding RNA expression profile in rabbit atria with atrial fibrillation: TCONS_00075467 modulates atrial electrical remodeling by sponging miR-328 to regulate CACNA1C. J. Mol. Cell. Cardiol. 2017, 108, 73–85, doi:10.1016/j.yjmcc.2017.05.009.
- Shen, C.; Kong, B.; Liu, Y.; Xiong, L.; Shuai, W.; Wang, G.; Quan, D.; Huang, H. YY1-induced upregulation of lncRNA KCNQ1OT1 regulates angiotensin II-induced atrial fibrillation by modulating miR-384b/CACNA1C axis. Biochem. Biophys. Res. Commun. 2018, 505, 134–140, doi:10.1016/j.bbrc.2018.09.064.
- Liu, J.; Li, Y.; Lin, B.; Sheng, Y.; Yang, L. HBL1 Is a Human Long Noncoding RNA that Modulates Cardiomyocyte Development from Pluripotent Stem Cells by Counteracting MIR1. Dev. Cell 2017, 42, 333–348, doi:10.1016/j.devcel.2017.07.023.
- Ruan, Z.; Sun, X.; Sheng, H.; Zhu, L. Long non-coding RNA expression profile in atrial fibrillation. Int. J. Clin. Exp. Pathol. 2015, 8, 8402–8410.
- Mei, B.; Liu, H.; Yang, S.; Liang, M.Y.; Yue, Y.; Huang, S.Q.; Hou, J.; Chen, G.X.; Wu, Z.K. Long non-coding RNA expression profile in permanent atrial fibrillation patients with rheumatic heart disease. Eur. Rev. Med. Pharmacol Sci. 2018, 22, 6940–6947, doi:10.26355/eurrev_201810_16165.
- Wirka, R.C.; Gore, S.; Van Wagoner, D.R.; Arking, D.E.; Lubitz, S.A.; Lunetta, K.L.; Benjamin, E.J.; Alonso, A.; Ellinor, P.T.; Barnard, J.; et al. A common connexin-40 gene promoter variant affects connexin-40 expression in human atria and is associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2011, 4, 87–93, doi:10.1161/CIRCEP.110.959726.
- Gegonne, A.; Devaiah, B.N.; Singer, D.S. TAF7: Traffic controller in transcription initiation. Transcription 2013, 4, 29–33, doi:10.4161/trns.22842.
- Wu, J.; Han, D.; Shi, R.; Chen, M.; Sun, J.; Tian, H.; Yan, Y. Identification of atrial fibrillation-associated lncRNAs in atria from patients with rheumatic mitral valve disease. Microsc. Res. Tech. 2019, 82, 1136–1144, doi:10.1002/jemt.23261.
- Ling, L.E.; Lee, W.C. Tgf-beta type I receptor (Alk5) kinase inhibitors in oncology. Curr. Pharm. Biotechnol. 2011, 12, 2190–2202, doi:10.2174/138920111798808257.
- Yu, X.J.; Zou, L.H.; Jin, J.H.; Xiao, F.; Li, L.; Liu, N.; Yang, J.F.; Zou, T. Long noncoding RNAs and novel inflammatory genes determined by RNA sequencing in human lymphocytes are up-regulated in permanent atrial fibrillation. Am. J. Transl. Res. 2017, 9, 2314–2326.
- Bettoni, M.; Zimmermann, M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation 2002, 105, 2753–2759, doi:10.1161/01.cir.0000018443.44005.d8.
- Zhao, Y.; Yuan, Y.; Qiu, C. Underexpression of CACNA1C Caused by Overexpression of microRNA-29a Underlies the Pathogenesis of Atrial Fibrillation. Med. Sci. Monit. 2016, 22, 2175–2181, doi:10.12659/msm.896191.
- Lu, Y.; Zhang, Y.; Wang, N.; Pan, Z.; Gao, X.; Zhang, F.; Zhang, Y.; Shan, H.; Luo, X.; Bai, Y.; et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 2010, 122, 2378–2387, doi:10.1161/CIRCULATIONAHA.110.958967.
- Nair, G.M.; Nery, P.B.; Redpath, C.J.; Birnie, D.H. The Role Of Renin Angiotensin System In Atrial Fibrillation. J. Atr. Fibrillation 2014, 6, 972, doi:10.4022/jafib.972.
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, J.C.; Grutzner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640, doi:10.1038/nature12943.
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011, 21, 579–587, doi:10.1038/cr.2010.163.
- Cyganek, L.; Tiburcy, M.; Sekeres, K.; Gerstenberg, K.; Bohnenberger, H.; Lenz, C.; Henze, S.; Stauske, M.; Salinas, G.; Zimmermann, W.H.; et al. Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 2018, 3, e99941, doi:10.1172/jci.insight.99941.
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179–194, doi:10.1016/j.stem.2017.07.003.
- Chodroff, R.A.; Goodstadt, L.; Sirey, T.M.; Oliver, P.L.; Davies, K.E.; Green, E.D.; Molnar, Z.; Ponting, C.P. Long noncoding RNA genes: Conservation of sequence and brain expression among diverse amniotes. Genome Biol. 2010, 11, R72, doi:10.1186/gb-2010-11-7-r72.
- Ng, S.Y.; Wong, C.K.; Tsang, S.Y. Differential gene expressions in atrial and ventricular myocytes: Insights into the road of applying embryonic stem cell-derived cardiomyocytes for future therapies. Am. J. Physiol. Cell Physiol. 2010, 299, C1234–1249, doi:10.1152/ajpcell.00402.2009.
- Del Alamo, J.C.; Lemons, D.; Serrano, R.; Savchenko, A.; Cerignoli, F.; Bodmer, R.; Mercola, M. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim. et Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 1717–1727, doi:10.1016/j.bbamcr.2016.03.003.
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410, doi:10.15252/emmm.201404757.
- Ghazizadeh, Z.; Kiviniemi, T.; Olafsson, S.; Plotnick, D.; Beerens, M.E.; Zhang, K.; Gillon, L.; Steinbaugh, M.J.; Barrera, V.; Sui, S.H.; et al. Metastable Atrial State Underlies the Primary Genetic Substrate for MYL4 Mutation-Associated Atrial Fibrillation. Circulation 2020, 141, 301–312, doi:10.1161/CIRCULATIONAHA.119.044268.
- Argenziano, M.; Lambers, E.; Hong, L.; Sridhar, A.; Zhang, M.; Chalazan, B.; Menon, A.; Savio-Galimberti, E.; Wu, J.C.; Rehman, J.; et al. Electrophysiologic Characterization of Calcium Handling in Human Induced Pluripotent Stem Cell-Derived Atrial Cardiomyocytes. Stem Cell Rep. 2018, 10, 1867–1878, doi:10.1016/j.stemcr.2018.04.005.
- Tanwar, V.; Bylund, J.B.; Hu, J.; Yan, J.; Walthall, J.M.; Mukherjee, A.; Heaton, W.H.; Wang, W.D.; Potet, F.; Rai, M.; et al. Gremlin 2 promotes differentiation of embryonic stem cells to atrial fate by activation of the JNK signaling pathway. Stem Cells 2014, 32, 1774–1788, doi:10.1002/stem.1703.
- Bylund, J.B.; Trinh, L.T.; Awgulewitsch, C.P.; Paik, D.T.; Jetter, C.; Jha, R.; Zhang, J.; Nolan, K.; Xu, C.; Thompson, T.B.; et al. Coordinated Proliferation and Differentiation of Human-Induced Pluripotent Stem Cell-Derived Cardiac Progenitor Cells Depend on Bone Morphogenetic Protein Signaling Regulation by GREMLIN 2. Stem Cells Dev. 2017, 26, 678–693, doi:10.1089/scd.2016.0226.
- Laksman, Z.; Wauchop, M.; Lin, E.; Protze, S.; Lee, J.; Yang, W.; Izaddoustdar, F.; Shafaattalab, S.; Gepstein, L.; Tibbits, G.F.; et al. Modeling Atrial Fibrillation using Human Embryonic Stem Cell-Derived Atrial Tissue. Sci. Rep. 2017, 7, 5268, doi:10.1038/s41598-017-05652-y.
- Benzoni, P.; Campostrini, G.; Landi, S.; Bertini, V.; Marchina, E.; Iascone, M.; Ahlberg, G.; Olesen, M.S.; Crescini, E.; Mora, C.; et al. Human iPSC modelling of a familial form of atrial fibrillation reveals a gain of function of If and ICaL in patient-derived cardiomyocytes. Cardiovasc. Res. 2020, 116, 1147–1160, doi:10.1093/cvr/cvz217.
- HONG, L.; Zhang, M.; Youn, S.-W.; Lambers, E.; Sridhar, A.; Menon, A.; Chalazan, B.; Wu, J.C.; Rehman, J.; Darbar, D. Abstract 407: Modeling Atrial Fibrillation in a Dish Using Atrial iPSC Derived Cardiomyocytes. Circ. Res. 2019, 125, A407–A407, doi:doi:10.1161/res.125.suppl_1.407.
- Girmatsion, Z.; Biliczki, P.; Bonauer, A.; Wimmer-Greinecker, G.; Scherer, M.; Moritz, A.; Bukowska, A.; Goette, A.; Nattel, S.; Hohnloser, S.H.; et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm. 2009, 6, 1802–1809, doi:10.1016/j.hrthm.2009.08.035.
- Jia, X.; Zheng, S.; Xie, X.; Zhang, Y.; Wang, W.; Wang, Z.; Zhang, Y.; Wang, J.; Gao, M.; Hou, Y. MicroRNA-1 accelerates the shortening of atrial effective refractory period by regulating KCNE1 and KCNB2 expression: An atrial tachypacing rabbit model. PLoS ONE 2013, 8, e85639, doi:10.1371/journal.pone.0085639.
- Celik, S.; Sadegh, M.K.; Morley, M.; Roselli, C.; Ellinor, P.T.; Cappola, T.; Smith, J.G.; Gidlof, O. Antisense regulation of atrial natriuretic peptide expression. JCI Insight 2019, 4, e130978, doi:10.1172/jci.insight.130978.

