 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jie Gu | + 2307 word(s) | 2307 | 2021-11-23 04:36:05 | | | |
| 2 | Catherine Yang | Meta information modification | 2307 | 2021-11-29 03:25:07 | | |
Video Upload Options
Ca2+ plays key roles in cells and decides the cell fate. During Ca2+ signaling mediates various cell deaths, such as necrosis, apoptosis, eryptosis as well as autophagy, which contribute to a series of kidney diseases, such as acute kidney injury (AKI), chronic kidney disease (CKD), renal ischemia/reperfusion (I/R) injury, autosomal dominant polycystic kidney disease (ADPKD), podocytopathy, and diabetic nephropathy. Importantly, there are complex Ca2+ flux networks between endoplasmic reticulum (ER), mitochondrial and lysosome, and Ca2+ signaling also links the crosstalk between various cell deaths and autophagy in kidney diseases.
1. Introduction
Through gradual evolutionary development, Calcium has become one of the most important metal elements in living organisms. With a wide variety of biological functions in living creatures, calcium ions (Ca 2+ ) are involved in almost every process from birth to death [1].
Ca 2+ is primarily stored in bones in the form of CaPO 3 (hydroxyapatite), where it plays a structural role and also can be dissolved to serve as a source of Ca 2 + in the blood [2]. In addition, Ca 2+ is a ubiquitous, multifunctional signaling molecule that controls a wide variety of life processes including muscle contraction, neuronal delivery, hormone secretion, organelle communication, cell movement, fertilization, and cell growth.
The kidney is an indispensable organ for maintaining body Ca 2+ homeostasis; the Ca 2+ signaling system in kidney cells regulates cellular processes and decides cell fate, including cell proliferation, apoptosis, necrosis and autophagy, all of which are associated with kidney disease [3][4]. Ca 2+ homeostasis has a certain influence on renal function and the occurrence and development of a series of nephropathies including acute kidney injury (AKI), chronic kidney disease (CKD), renal ischemia/reperfusion (I/R) injury, autosomal dominant polycystic kidney disease (ADPKD), podocytopathy, and diabetic nephropathy, which will be discussed in the following sections.
2. Relationship between Ca2+ Signaling and Various Forms of Cell Death in Kidney Cells
Apoptosis, or autonomous programmed cell death, is an important process for the maintenance of the stability of the internal environment, and helps an organism better adapt to its living environment. In kidney injury, apoptosis usually occurs in association with necrosis, and Ca 2+ signaling acts as an important regulator of apoptosis. Ca 2+ overload leads to necrotic or apoptotic death in renal ischemia–reperfusion (I/R) injury [5]. Sustained ER Ca 2+ release induces ER stress and oxidative stress and leads to apoptosis in glomerular mesangial cells, which contributes to the progression of CKD [6]. CaSR is a pleiotropic receptor capable of regulating Ca 2+ homeostasis, and plays important roles in kidney cells and cancers [7]. Activation of adiponectin receptors by AdipoRon and activation of CaSR by cinacalcet increases intracellular Ca 2+ levels, which inhibits apoptosis in the kidneys induced by high glucose and ameliorates glomerular endothelial cell and podocyte injury in type 2 diabetes-associated diabetic nephropathy [8][9]. This suggests that regulation of cytosolic Ca 2+ can control the apoptosis of kidney cells.
Macroautophagy/autophagy, an evolutionarily conserved process in eukaryotes, plays an important role in intracellular material recycling. In the process of autophagy, some damaged organelles and harmful proteins are wrapped by the autophagosomes with a double membrane structure, then sent into lysosomes or vacuoles for degradation and reuse [10]. Ca 2+ , as an important messenger molecule that regulates cell death, is also involved in the regulation of autophagy [11].
It has been suggested that intracellular Ca 2+ signaling mediates autophagy in renal tubular cells. An in vivo study has shown that the key regulator of the autophagy pathway, mTOR, is involved in tubular repair after AKI [12]. In conditionally immortalized proximal tubular epithelial cells (ciPTEC) generated from an ADPKD1 patient, activation of CaSR increased intracellular Ca 2+ release and decreased mTOR activity [13]. Increased Ca 2+ influx in renal proximal tubular cells inhibits mTOR-dependent autophagy, thereby rendering cells more susceptible to death [14]. This indicates that the mTOR-dependent autophagy regulated by intracellular Ca 2+ release or Ca 2+ influx controls the development of kidney disease.
The canonical transient receptor potential channel 6 (TRPC6), a major Ca 2+ influx channel in renal cells, plays an important role in such renal diseases as diabetic nephropathy, immune-mediated kidney disease, renal fibrosis, glomerular disease and CKD [15]. An in vitro study in renal proximal tubular cells showed that the cytoprotective role of autophagy was suppressed by TRPC6-mediated Ca 2+ influx [14]. The same study showed that TRPC6 knockout promoted autophagy flux and alleviated tubular apoptosis upon renal I/R, a major cause of AKI [16]. The transient receptor potential non-selective cation channel, subfamily M, member 3 (TRPM3) is another Ca 2+ channel that conducts Ca 2+ flux to regulating autophagy. Increased expression of TRPM3 leads to Ca 2+ influx and stimulates autophagy through the CAMKK2/AMPK/ULK1 pathway, which promotes the growth of clear cell renal cell carcinoma (ccRCC) [17]. These results suggest that Ca 2+ channels on the cell membrane mediate Ca 2+ influx and suppress autophagy, which may contribute to kidney injury or diseases.
3. Ca2+ Signaling Links Cell Death and Autophagy in Kidney Cells
The relation of cell death and autophagy is complex and occasionally contradictory, but it is critical to cell fate. Intriguingly, Ca 2+ signaling acts as a bridge linking these two types of cellular activities [18][19][20][21][22]. Ca 2+ promote cell proliferation and survival through release of IP3R by the ER; Ca 2+ is subsequently transferred to mitochondria to activate mitochondrial metabolism. Mitochondrial Ca 2+ homeostasis dysfunction results in mitochondrial degradation by autophagy via activation of AMPK. Mitochondrial Ca 2+ overload causes production of reactive oxygen species (ROS) and release of cytochrome c, which eventually leads to cell apoptosis [11][23][24]. Therefore, Ca 2+ signaling and Ca 2+ subcellular homeostasis may determine the balance between cell survival, apoptosis and autophagy.
In the renal fibrosis model of unilateral ureteral obstruction (UUO) in mice, persistent autophagy in kidney proximal tubules was observed. Pharmacological inhibition of autophagy and kidney proximal tubule-specific knockout of autophagy-related 7 (PT-ATG7 KO) suppressed tubular atrophy, apoptosis, nephron loss, and interstitial macrophage infiltration in these mice [25]. This suggests that persistent induction of autophagy in kidney proximal tubules promotes renal interstitial fibrosis during UUO. In addition, influx of extracellular Ca 2+ triggered by Trichokonin VI, an antimicrobial peptide, induces autophagy and apoptosis in hepatocellular carcinoma cells. Moreover, siRNA knockdown of autophagy related gene (ATG5) reduces cell apoptosis [19]. This shows that Cd induces the mitochondrial-derived autophagic cell death of hepatocytes in a dose-dependent manner. By suppressing Cd-induced autophagic cell death, melatonin has a hepatoprotective effect in Cd-exposed mice [26]. In mouse spleen and human B cells, Cd induces vacuole membrane protein 1 (VMP1)-mediated autophagy via elevation of intracellular Ca 2 + , which contributes to apoptosis [27]. In RAW264.7 mouse monocytes, Cd induced autophagy and ER-mediated apoptosis; however, pharmacological and genetic inhibition of autophagy suppressed Cd-induced apoptosis. Moreover, treatment with Ca 2+ chelators completely restored cell viability and inhibited Cd-induced apoptosis and autophagy [28]. In porcine kidney cell LLC-PK1, the autophagy mediator calpain induced necrosis before apoptosis by increasing intracellular Ca 2+ levels in high-glucose conditions [29]. In addition, Ca 2+ also plays important roles in ferroptosis [30], a type of autophagy-dependent cell death [31] which has recently been shown to have implications in diverse kidney diseases [32]. These studies suggest that intracellular Ca 2+ signaling-mediated autophagy may promote cell death and contribute to kidney disease.
It has been shown that high glucose promotes autophagy flux in podocyte cultures and induces LC3B-II expression in podocytes in diabetic mice. Specifically, deletion of ATG5 in podocytes resulted in accelerated diabetes-induced podocytopathy with a leaky glomerular filtration barrier and glomerulosclerosis. Furthermore, the endothelial-specific deletion of ATG5 also resulted in capillary rarefaction and accelerated diabetic nephropathy. Thus, endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis [33]. In mouse mesangial cells (MES-13), Cd induced autophagy and apoptosis by inducing ER Ca 2+ release through IP3R [34]. In addition, Cd induced the expression of LC3B-II but reduced the expression of sequestosome-1 (p62) in rat mesangial cells. When autophagy is disrupted either by gene knockout or RNA silencing, cell viability is decreased and increased pro-caspase-3 cleavage indicates the initiation of apoptotic cell death [35]. These results suggest that induced autophagy may protect against nephrotoxicity.
However, initial autophagic protection will switch to disruption of autophagic flux, resulting in cell death in renal cells [36]. In other words, inhibition of autophagy contributes to cell death. The autophagy inhibitor 3-methyladenine exacerbates Cd-caused germ cell apoptosis, which is relieved by the autophagy inducer rapamycin. More importantly, loss of ATG5 in Sertoli cells aggravates Cd-triggered germ cell apoptosis. This suggests that autophagy in Sertoli cells protects against Cd-induced germ cell apoptosis in mouse testes [37]. In human placental trophoblasts and mouse placenta, it has also been shown that activation of autophagy inhibits Cd-triggered apoptosis [38]. As an important excretory organ, the kidney is the main accumulation target of toxins such as heavy metals [39][40]. Previous studies have shown that Cd induces kidney injury and apoptosis via long-term inhibition of autophagy flux [41]. In vitro studies also show that inhibition of autophagy flux can aggravate cell apoptosis; Ca 2+ signaling may link these two cell activities. In mouse renal tubular cells, Cd-inhibited autophagy flux aggravated apoptosis by inducing elevation of Ca 2+ level [42]. In primary rat proximal tubular cells, Cd and lead (Pb)-inhibited autophagic degradation aggravated apoptotic death [43], which could be due to the redistribution of subcellular Ca 2+ between the ER, cytosol and mitochondria [44][45]. Activation of CaSR can promote cell proliferation, and protects against Cd-induced renal tubular cell apoptosis through competing PLC-IP3-Ca 2+ signaling [42]. Restoring the Ca 2+ -mediated autophagy process can protect against heavy metal-induced renal cell cytotoxicity and kidney injury [41][46][47].
4. Targeted Ca2+ Signaling for Therapy of Kidney Diseases
As described above, the Ca 2+ microdomains regulating apoptosis, necrosis and autophagy contribute to the development of kidney disease. Under these conditions, Ca 2+ signaling determines the fate of kidney cells and the progression of disease. In view of this, therapeutic strategies have to consider whether to target a specific microdomain of Ca 2+ signaling in kidney disease.
Firstly, ER Ca 2+ signaling-mediated apoptotic pathways in kidney disease could be considered as potential therapeutic targets ( Figure 1 ). Regulating ER stress in kidney cells may provide a therapeutic target in acute kidney injury triggered by renal ischemia reperfusion and cisplatin nephrotoxicity [48]. Pretreatment with trans-4,5-dihydroxy-1,2-dithiane (DTTox) enhances expression of the ER stress markers glucose-regulated protein 78 (GRP78) and GRP94 and protects against increased cellular Ca 2+ levels and cell death in LLC-PK1 renal epithelial cells [49], as well as protecting the kidneys from nephrotoxic injury in vivo [50]. Treatment with tauroursodeoxycholic acid (TUDCA) prevents advanced glycation end product (AGE)-induced apoptosis of mouse podocytes in diabetic nephropathy by blocking an ER Ca 2+ release-mediated apoptotic pathway [51]. By knocking down STIM1 levels, it can reduce Ca 2+ release and restore intracellular Ca 2+ homeostasis, which decreases PC2 protein levels in PC1-null proximal tubule cells and inhibits cyst growth in ADPKD [52].
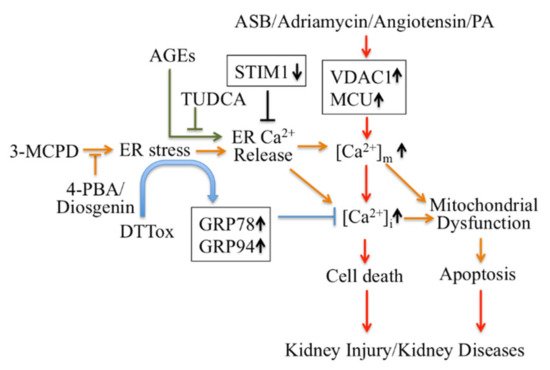
Secondly, modulators targeting mitochondrial Ca 2+ mediate apoptotic pathways and may also be treatments for kidney disease therapy ( Figure 1 ). Inhibition of ER stress by 4-phenylbutyric acid (4-PBA) and Diosgenin mitigates ER-associated mitochondrial apoptosis by maintaining Ca 2+ homeostasis and mitochondrial dynamics, which ameliorates 3-MCPD-induced kidney injury [53][54]. In addition, 4-PBA also decreases Porcine Circovirus Type 2 (PCV2) infection-induced apoptosis by decreasing the cytosolic and mitochondrial Ca 2+ load in porcine kidney PK-15 cells [55]. Mitochondrial outer membrane-located voltage-dependent anion channel (VDAC1) acts as gatekeeper for Ca 2+ distribution between the mitochondria, cytosol and ER. Proteomic identification showed VDAC1 to be one the mitochondrial targets involved in andrographolide sodium bisulfite (ASB)-induced nephrotoxicity in a rat model [56]. Palmitic acid (PA) induced apoptosis through disruption of calcium homeostasis in mice podocytes [57][58]. In addition, Palmitic acid induced a continual increase in autophagy, ER stress, and apoptosis in primary cultured proximal tubular cells, and markedly upregulated VDAC1, which is associated with mitochondrial damage in HK-2 cells and may contribute to tubular injuries in obesity-related kidney disease [59]. In Adriamycin- or angiotensin II-treated rats, expression of VDAC1 and mitochondrial calcium uniporter (MCU) were upregulated; these mediate podocyte apoptosis by facilitating mitochondrial Ca 2+ overload. However, MCU inhibitors can protect podocytes from apoptosis and proteinuria induced by Adriamycin or angiotensin II [60]. This suggests that regulating mitochondrial Ca 2+ may also be a potential target for some stress-induced nephropathies.
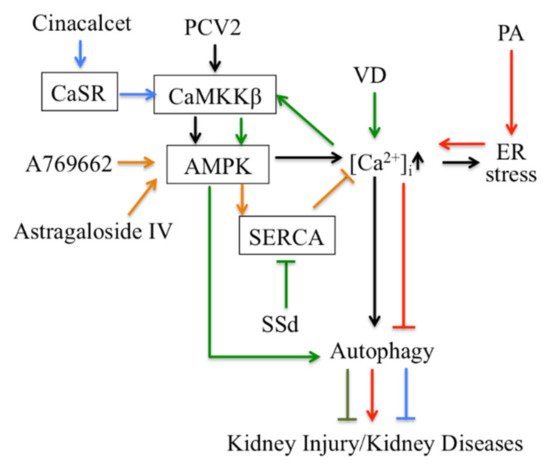
Thirdly, Ca 2+ signaling-mediated autophagy provides new therapeutic ways to treat kidney disease ( Figure 2 ). PCV2 induces autophagy via the CaMKKβ-AMPK pathway in pig kidney PK-15 cells by increasing cytosolic Ca 2+ [61]. Activation of vitamin D receptors can restore defective autophagy through the Ca 2+ -CAMKKβ-AMPK pathway in renal tubular epithelial cells in streptozotocin-induced diabetic mice [62]. Ca 2+ signaling is involved in AMPK-mediated autophagy, and plays a role in coordinating cellular survival and kidney function. A selective activator of AMPK (A769662) reduced intracellular Ca 2+ by activation of SERCA in vascular smooth muscle, which produces vasodilation in human intrarenal arteries [63]. This suggests that activation of AMPK and SERCA might be therapeutic targets in kidney diseases. Podocyte apoptosis induced by diabetes or high glucose and progression of diabetic nephropathy are prevented by astragaloside IV, which attenuates SERCA2-dependent ER stress and induces AMPKα-promoted autophagy [64]. Saikosaponin-d (SSd), a SERCA inhibitor, suppresses excess ER Ca 2+ reuptake and cell proliferation in ADPKD cells by inducing autophagy through the CaMKKβ-AMPK-mTOR signaling pathway, which indicates that SSd might be a potential treatment for ADPKD and that SERCA might be a novel target for ADPKD therapy [65]. It has been reported that AMPK activation restored the defective autophagy in high glucose-induced HK-2 cells [62]. Activators of CaSR such as cinacalcet have renoprotective effects in high glucose-treated human glomerular endothelial cells, murine podocytes and diabetic mice. Cinacalcet decreases oxidative stress and apoptosis and increases autophagy by increasing intracellular Ca 2+ level through activation of the CaMKKβ-LKB1-AMPK pathway in glomerular endothelial cells and podocytes in the kidney [9]. Taken together, Ca 2+ signaling-mediated autophagy might be a potential target in therapy for metabolic disease-associated kidney diseases.
References
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721.
- Pozzan, T.; Rizzuto, R.; Volpe, P.; Meldolesi, J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994, 74, 595–636.
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529.
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058.
- Pittas, K.; Vrachatis, D.A.; Angelidis, C.; Tsoucala, S.; Giannopoulos, G.; Deftereos, S. The Role of Calcium Handling Mechanisms in Reperfusion Injury. Curr. Pharm. Des. 2018, 24, 4077–4089.
- Mehta, N.; Gava, A.L.; Zhang, D.; Gao, B.; Krepinsky, J.C. Follistatin Protects Against Glomerular Mesangial Cell Apoptosis and Oxidative Stress to Ameliorate Chronic Kidney Disease. Antioxid. Redox Signal. 2019, 31, 551–571.
- Tuffour, A.; Kosiba, A.A.; Zhang, Y.; Peprah, F.A.; Gu, J.; Shi, H. Role of the calcium-sensing receptor (CaSR) in cancer metastasis to bone: Identifying a potential therapeutic target. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188528.
- Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Yoon, H.E.; Shin, S.J.; Choi, B.S.; Kim, Y.S.; Chang, Y.S.; Park, C.W. The Adiponectin Receptor Agonist AdipoRon Ameliorates Diabetic Nephropathy in a Model of Type 2 Diabetes. J. Am. Soc. Nephrol. JASN 2018, 29, 1108–1127.
- Lim, J.H.; Kim, H.W.; Kim, M.Y.; Kim, T.W.; Kim, E.N.; Kim, Y.; Chung, S.; Kim, Y.S.; Choi, B.S.; Kim, Y.S.; et al. Cinacalcet-mediated activation of the CaMKKβ-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death Dis. 2018, 9, 270.
- Zhang, Y.; Li, K.; Kong, A.; Zhou, Y.; Chen, D.; Gu, J.; Shi, H. Dysregulation of autophagy acts as a pathogenic mechanism of non-alcoholic fatty liver disease (NAFLD) induced by common environmental pollutants. Ecotoxicol. Environ. Saf. 2021, 217, 112256.
- La Rovere, R.M.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87.
- Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury. Semin. Nephrol. 2014, 34, 17–26.
- Di Mise, A.; Tamma, G.; Ranieri, M.; Centrone, M.; van den Heuvel, L.; Mekahli, D.; Levtchenko, E.N.; Valenti, G. Activation of Calcium-Sensing Receptor increases intracellular calcium and decreases cAMP and mTOR in PKD1 deficient cells. Sci. Rep. 2018, 8, 5704.
- Hou, X.; Xiao, H.; Zhang, Y.; Zeng, X.; Huang, M.; Chen, X.; Birnbaumer, L.; Liao, Y. Transient receptor potential channel 6 knockdown prevents apoptosis of renal tubular epithelial cells upon oxidative stress via autophagy activation. Cell Death Dis. 2018, 9, 1015.
- Hall, G.; Wang, L.; Spurney, R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells 2019, 9, 44.
- Hou, X.; Huang, M.; Zeng, X.; Zhang, Y.; Sun, A.; Wu, Q.; Zhu, L.; Zhao, H.; Liao, Y. The Role of TRPC6 in Renal Ischemia/Reperfusion and Cellular Hypoxia/Reoxygenation Injuries. Front. Mol. Biosci. 2021, 8, 698975.
- Hall, D.P.; Cost, N.G.; Hegde, S.; Kellner, E.; Mikhaylova, O.; Stratton, Y.; Ehmer, B.; Abplanalp, W.A.; Pandey, R.; Biesiada, J.; et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell 2014, 26, 738–753.
- Smaili, S.S.; Pereira, G.J.; Costa, M.M.; Rocha, K.K.; Rodrigues, L.; do Carmo, L.G.; Hirata, H.; Hsu, Y.T. The role of calcium stores in apoptosis and autophagy. Curr. Mol. Med. 2013, 13, 252–265.
- Shi, M.; Zhang, T.; Sun, L.; Luo, Y.; Liu, D.H.; Xie, S.T.; Song, X.Y.; Wang, G.F.; Chen, X.L.; Zhou, B.C.; et al. Calpain, Atg5 and Bak play important roles in the crosstalk between apoptosis and autophagy induced by influx of extracellular calcium. Apoptosis Int. J. Program. Cell Death 2013, 18, 435–451.
- Mammano, F.; Bortolozzi, M. Ca(2+) signaling, apoptosis and autophagy in the developing cochlea: Milestones to hearing acquisition. Cell Calcium 2018, 70, 117–126.
- Zhou, X.; Hao, W.; Shi, H.; Hou, Y.; Xu, Q. Calcium homeostasis disruption—A bridge connecting cadmium-induced apoptosis, autophagy and tumorigenesis. Oncol. Res. Treat. 2015, 38, 311–315.
- Kosiba, A.A.; Wang, Y.; Chen, D.; Wong, C.K.C.; Gu, J.; Shi, H. The roles of calcium-sensing receptor (CaSR) in heavy metals-induced nephrotoxicity. Life Sci. 2020, 242, 117183.
- Decuypere, J.P.; Monaco, G.; Bultynck, G.; Missiaen, L.; De Smedt, H.; Parys, J.B. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta 2011, 1813, 1003–1013.
- Wacquier, B.; Combettes, L.; Van Nhieu, G.T.; Dupont, G. Interplay Between Intracellular Ca(2+) Oscillations and Ca(2+)-stimulated Mitochondrial Metabolism. Sci. Rep. 2016, 6, 19316.
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998.
- Pi, H.; Xu, S.; Reiter, R.J.; Guo, P.; Zhang, L.; Li, Y.; Li, M.; Cao, Z.; Tian, L.; Xie, J.; et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy 2015, 11, 1037–1051.
- Gu, J.; Wang, Y.; Liu, Y.; Shi, M.; Yin, L.; Hou, Y.; Zhou, Y.; Wong, C.K.C.; Chen, D.; Guo, Z.; et al. Inhibition of Autophagy Alleviates Cadmium-Induced Mouse Spleen and Human B Cells Apoptosis. Toxicol. Sci. 2019, 170, 109–122.
- So, K.Y.; Lee, B.H.; Oh, S.H. The critical role of autophagy in cadmium-induced immunosuppression regulated by endoplasmic reticulum stress-mediated calpain activation in RAW264.7 mouse monocytes. Toxicology 2018, 393, 15–25.
- Harwood, S.M.; Allen, D.A.; Raftery, M.J.; Yaqoob, M.M. High glucose initiates calpain-induced necrosis before apoptosis in LLC-PK1 cells. Kidney Int. 2007, 71, 655–663.
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca(2+) in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55.
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100.
- Kim, S.; Kang, S.W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.H.; Kim, G.; Lee, P.; et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160.
- Lenoir, O.; Jasiek, M.; Hénique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145.
- Wang, S.H.; Shih, Y.L.; Ko, W.C.; Wei, Y.H.; Shih, C.M. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell. Mol. life Sci. CMLS 2008, 65, 3640–3652.
- Fujishiro, H.; Liu, Y.; Ahmadi, B.; Templeton, D.M. Protective effect of cadmium-induced autophagy in rat renal mesangial cells. Arch. Toxicol. 2018, 92, 619–631.
- Lee, W.K.; Probst, S.; Santoyo-Sánchez, M.P.; Al-Hamdani, W.; Diebels, I.; von Sivers, J.K.; Kerek, E.; Prenner, E.J.; Thévenod, F. Initial autophagic protection switches to disruption of autophagic flux by lysosomal instability during cadmium stress accrual in renal NRK-52E cells. Arch. Toxicol. 2017, 91, 3225–3245.
- Zhou, G.X.; Zhu, H.L.; Shi, X.T.; Nan, Y.; Liu, W.B.; Dai, L.M.; Xiong, Y.W.; Yi, S.J.; Cao, X.L.; Xu, D.X.; et al. Autophagy in Sertoli cell protects against environmental cadmium-induced germ cell apoptosis in mouse testes. Environ. Pollut. 2021, 270, 116241.
- Zhu, H.L.; Xu, X.F.; Shi, X.T.; Feng, Y.J.; Xiong, Y.W.; Nan, Y.; Zhang, C.; Gao, L.; Chen, Y.H.; Xu, D.X.; et al. Activation of autophagy inhibits cadmium-triggered apoptosis in human placental trophoblasts and mouse placenta. Environ. Pollut. 2019, 254, 112991.
- Shi, H.; Sun, X.; Kong, A.; Ma, H.; Xie, Y.; Cheng, D.; Wong, C.K.C.; Zhou, Y.; Gu, J. Cadmium induces epithelial-mesenchymal transition and migration of renal cancer cells by increasing PGE2 through a cAMP/PKA-COX2 dependent mechanism. Ecotoxicol. Environ. Saf. 2021, 207, 111480.
- Sun, X.; Wang, Y.; Jiang, T.; Yuan, X.; Ren, Z.; Tuffour, A.; Liu, H.; Zhou, Y.; Gu, J.; Shi, H. Nephrotoxicity Profile of Cadmium Revealed by Proteomics in Mouse Kidney. Biol. Trace Elem. Res. 2021, 199, 1929–1940.
- Gu, J.; Ren, Z.; Zhao, J.; Peprah, F.A.; Xie, Y.; Cheng, D.; Wang, Y.; Liu, H.; Chu Wong, C.K.; Zhou, Y.; et al. Calcimimetic compound NPS R-467 protects against chronic cadmium-induced mouse kidney injury by restoring autophagy process. Ecotoxicol. Environ. Saf. 2020, 189, 110052.
- Gu, J.; Dai, S.; Liu, Y.; Liu, H.; Zhang, Y.; Ji, X.; Yu, F.; Zhou, Y.; Chen, L.; Tse, W.K.F.; et al. Activation of Ca(2+)-sensing receptor as a protective pathway to reduce Cadmium-induced cytotoxicity in renal proximal tubular cells. Sci. Rep. 2018, 8, 1092.
- Chu, B.X.; Fan, R.F.; Lin, S.Q.; Yang, D.B.; Wang, Z.Y.; Wang, L. Interplay between autophagy and apoptosis in lead(II)-induced cytotoxicity of primary rat proximal tubular cells. J. Inorg. Biochem. 2018, 182, 184–193.
- Wang, H.; Wang, Z.K.; Jiao, P.; Zhou, X.P.; Yang, D.B.; Wang, Z.Y.; Wang, L. Redistribution of subcellular calcium and its effect on apoptosis in primary cultures of rat proximal tubular cells exposed to lead. Toxicology 2015, 333, 137–146.
- Liu, F.; Li, Z.F.; Wang, Z.Y.; Wang, L. Role of subcellular calcium redistribution in regulating apoptosis and autophagy in cadmium-exposed primary rat proximal tubular cells. J. Inorg. Biochem. 2016, 164, 99–109.
- Liu, F.; Wang, X.Y.; Zhou, X.P.; Liu, Z.P.; Song, X.B.; Wang, Z.Y.; Wang, L. Cadmium disrupts autophagic flux by inhibiting cytosolic Ca(2+)-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology 2017, 383, 13–23.
- Wang, X.Y.; Yang, H.; Wang, M.G.; Yang, D.B.; Wang, Z.Y.; Wang, L. Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis. 2017, 8, e3099.
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390.
- Liu, H.; Bowes, R.C., 3rd; van de Water, B.; Sillence, C.; Nagelkerke, J.F.; Stevens, J.L. Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J. Biol. Chem. 1997, 272, 21751–21759.
- Asmellash, S.; Stevens, J.L.; Ichimura, T. Modulating the endoplasmic reticulum stress response with trans-4,5-dihydroxy-1,2-dithiane prevents chemically induced renal injury in vivo. Toxicol. Sci. 2005, 88, 576–584.
- Chen, Y.; Liu, C.P.; Xu, K.F.; Mao, X.D.; Lu, Y.B.; Fang, L.; Yang, J.W.; Liu, C. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am. J. Nephrol. 2008, 28, 1014–1022.
- Yanda, M.K.; Liu, Q.; Cebotaru, V.; Guggino, W.B.; Cebotaru, L. Role of calcium in adult onset polycystic kidney disease. Cell. Signal. 2019, 53, 140–150.
- Zhong, Y.; Jin, C.; Han, J.; Zhu, J.; Liu, Q.; Sun, D.; Xia, X.; Peng, X. Inhibition of ER stress attenuates kidney injury and apoptosis induced by 3-MCPD via regulating mitochondrial fission/fusion and Ca(2+) homeostasis. Cell Biol. Toxicol. 2021, 37, 795–809.
- Zhong, Y.; Jin, C.; Han, J.; Zhu, J.; Liu, Q.; Sun, D.; Xia, X.; Zhang, Y.; Peng, X. Diosgenin Protects Against Kidney Injury and Mitochondrial Apoptosis Induced by 3-MCPD Through the Regulation of ER Stress, Ca(2+) Homeostasis, and Bcl2 Expression. Mol. Nutr. Food Res. 2021, 65, e2001202.
- Zhang, Y.; Sun, R.; Geng, S.; Shan, Y.; Li, X.; Fang, W. Porcine Circovirus Type 2 Induces ORF3-Independent Mitochondrial Apoptosis via PERK Activation and Elevation of Cytosolic Calcium. J. Virol. 2019, 93, e01784-18.
- Xing, W.M.; Yuan, T.J.; Xu, J.D.; Gu, L.L.; Liang, P.; Lu, H. Proteomic identification of mitochondrial targets involved in andrographolide sodium bisulfite-induced nephrotoxicity in a rat model. Environ. Toxicol. Pharmacol. 2015, 40, 592–599.
- Yuan, Z.; Cao, A.; Liu, H.; Guo, H.; Zang, Y.; Wang, Y.; Wang, Y.; Wang, H.; Yin, P.; Peng, W. Calcium Uptake via Mitochondrial Uniporter Contributes to Palmitic Acid-Induced Apoptosis in Mouse Podocytes. J. Cell. Biochem. 2017, 118, 2809–2818.
- Zang, Y.; Liu, S.; Cao, A.; Shan, X.; Deng, W.; Li, Z.; Wang, H.; Wang, Y.; Wang, L.; Peng, W. Astragaloside IV inhibits palmitic acid-induced apoptosis through regulation of calcium homeostasis in mice podocytes. Mol. Biol. Rep. 2021, 48, 1453–1464.
- Kong, Y.; Zhao, X.; Qiu, M.; Lin, Y.; Feng, P.; Li, S.; Liang, B.; Zhu, Q.; Huang, H.; Li, C.; et al. Tubular Mas receptor mediates lipid-induced kidney injury. Cell Death Dis. 2021, 12, 110.
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. IP(3)R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140.
- Gu, Y.; Qi, B.; Zhou, Y.; Jiang, X.; Zhang, X.; Li, X.; Fang, W. Porcine Circovirus Type 2 Activates CaMMKβ to Initiate Autophagy in PK-15 Cells by Increasing Cytosolic Calcium. Viruses 2016, 8, 135.
- Li, A.; Yi, B.; Han, H.; Yang, S.; Hu, Z.; Zheng, L.; Wang, J.; Liao, Q.; Zhang, H. Vitamin D-VDR (vitamin D receptor) regulates defective autophagy in renal tubular epithelial cell in streptozotocin-induced diabetic mice via the AMPK pathway. Autophagy 2021, 1–14.
- Rodríguez, C.; Contreras, C.; Sáenz-Medina, J.; Muñoz, M.; Corbacho, C.; Carballido, J.; García-Sacristán, A.; Hernandez, M.; López, M.; Rivera, L.; et al. Activation of the AMP-related kinase (AMPK) induces renal vasodilatation and downregulates Nox-derived reactive oxygen species (ROS) generation. Redox Biol. 2020, 34, 101575.
- Guo, H.; Wang, Y.; Zhang, X.; Zang, Y.; Zhang, Y.; Wang, L.; Wang, H.; Wang, Y.; Cao, A.; Peng, W. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKα-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci. Rep. 2017, 7, 6852.
- Shi, W.; Xu, D.; Gu, J.; Xue, C.; Yang, B.; Fu, L.; Song, S.; Liu, D.; Zhou, W.; Lv, J.; et al. Saikosaponin-d inhibits proliferation by up-regulating autophagy via the CaMKKβ-AMPK-mTOR pathway in ADPKD cells. Mol. Cell. Biochem. 2018, 449, 219–226.

