 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Birgitt Schuele | + 4335 word(s) | 4335 | 2020-08-13 07:38:19 | | | |
| 2 | Bruce Ren | Meta information modification | 4335 | 2020-08-24 10:04:27 | | | | |
| 3 | Bruce Ren | Meta information modification | 4335 | 2020-08-24 10:04:55 | | | | |
| 4 | Bruce Ren | Meta information modification | 4335 | 2020-08-24 10:05:18 | | |
Video Upload Options
Neurodevelopmental and late-onset neurodegenerative disorders present as separate entities that are clinically and neuropathologically quite distinct. However, recent evidence has highlighted surprising commonalities and converging features at the clinical, genomic, and molecular level between these two disease spectra. This is particularly striking in the context of autism spectrum disorder (ASD) and Parkinson’s disease (PD). Genetic causes and risk factors play a central role in disease pathophysiology and enable the identification of overlapping mechanisms and pathways. Several genes and genomic regions are highlighted, including SNCA (alpha-synuclein), PARK2 (parkin RBR E3 ubiquitin protein ligase), chromosome 22q11 deletion/DiGeorge region, and FMR1 (fragile X mental retardation 1) repeat expansion, which influence the development of both ASD and PD, with converging features related to synaptic function and neurogenesis. Both PD and ASD display alterations and impairments at the synaptic level, representing early and key disease phenotypes, which support the hypothesis of converging mechanisms between the two types of diseases. Therefore, understanding the underlying molecular mechanisms might inform on common targets and therapeutic approaches.
Neurodevelopmental and late-onset neurodegenerative disorders present as separate entities that are clinically and neuropathologically quite distinct. However, recent evidence has highlighted surprising commonalities and converging features at the clinical, genomic, and molecular level between these two disease spectra. This is particularly striking in the context of autism spectrum disorder (ASD) and Parkinson’s disease (PD). Genetic causes and risk factors play a central role in disease pathophysiology and enable the identification of overlapping mechanisms and pathways.
1. Introduction of Clinical Aspects of Parkinson’s Disease (PD) and Autism Spectrum Disorders (ASDs)
Early onset neurodevelopmental and late-onset neurodegenerative disorders present as entities that appear quite distinct. However, recent evidence has uncovered surprising commonalities and converging features at the clinical, genomic, and molecular level between these two disease spectra and, in particular, between ASD and PD.
PD is a progressive neurodegenerative disorder characterized by cardinal motor features, including resting tremor, rigidity, postural instability, and bradykinesia. In addition, PD may also present with a wide range of non-motor symptoms, such as autonomic dysfunction, cognitive impairment, sleep disturbances, and neuropsychiatric symptoms, including depression, anxiety, and repetitive or obsessive–compulsive behaviors [1][2][3]. The prevalence of PD is about 1% over the age of 65. More men than women are diagnosed with sporadic PD and this ratio increases with age; while the incidence ratio is comparable in males and females under 50 years (male:female ratio < 1.2), it increases to 1.6 above 80 years [4]. The neuropathological changes that underlie the Lewy body disease (LBD) spectrum include PD, dementia with Lewy bodies (DLB), and Parkinson’s disease dementia (PDD) [5]. These diseases are characterized by the accumulation of intracellular protein inclusions immunoreactive for several proteins but primarily comprising alpha-synuclein (α-syn) and α-syn phosphorylated at position S129 [6][7][8]. Neurons most vulnerable for LB pathology are projection neurons with disproportionately long axons and poor myelination—a key example being dopaminergic neurons projecting from the substantia nigra (SN) into the striatum [9][10]. Both genetic factors and environmental exposure build the basis for defining the causality of LBD [11][12][13][14][15][16][17]. A number of Mendelian forms of PD have been described in the last two decades in addition to risk genes identified through candidate gene studies and genome wide-association studies (GWAS) [18][19]. One of the largest GWAS studies to date reports 90 independent genome-wide significant risk signals across 78 genomic regions genotyping 7.8 million single nucleotide polymorphisms (SNPs) in 37,700 cases, 18,600 proxy cases, and 1.4 million controls [20]. Interestingly, tissue-specific expression enrichment analyses suggest that PD loci are heavily brain enriched, e.g., in neurons from the SN, globus pallidus (GP), thalamus, posterior cortex (PC), frontal cortex (FC), hippocampus (HC), and entopeduncular nucleus (ENT, internal part of the globus pallidus) [20]. Disease mechanisms and phenotypes related to PD range from toxic protein aggregation, impairment of mitochondrial and lysosomal/autophagy function, disruption of vesicle and endocytosis, and dopamine metabolism. Head injury and lifestyle factors as well as exposure to environmental toxicants and contaminants, including pesticides, solvents, or metals, among other pollutants, have been found to increase risk for PD and have been reproduced and validated in animal models [16][17][21].
Autism spectrum disorder (ASD), on the other hand, is a multifactorial neurodevelopmental disorder with a prevalence of 1.0–2.6%, presenting with three core symptoms: Impairments in social interaction; communication impairments; and restricted, repetitive, and stereotyped patterns of behaviors [22][23]. Additional features that can accompany ASD are motor abnormalities, gastrointestinal problems, epilepsy, intellectual disability, or sleep disorders [24]. Males are more frequently diagnosed with ASD than females, with a prevalence ratio of about 4:1 (male:female) [25][26]. Key neuroanatomical features of ASD are early brain overgrowth during toddler years, which disappears between 5–6 years of age [27]. Regions affected include the fronto-temporal and frontoparietal regions, the amygdala-hippocampal complex, cerebellum, basal ganglia, and anterior and posterior cingulate regions [28]. Microscopically, there are a decreased perikaryal size in layers III and V of the fusiform gyrus of the cortex, altered cell distribution of cortical layers with a less distinct lamination in the architecture, and abnormal neuronal morphology of von Economo neurons (VENs) [29] in the fronto-insular cortex and anterior limbic area with corkscrew dendrites and swollen soma [30]. The time period of early brain overgrowth suggests that neurodevelopmental processes are affected, including synaptogenesis, axonal and synaptic pruning, and myelination, leading to an altered ultrastructure of synapses and structural connectivity [31]. Clinical genetic studies identified more than 100 ASD candidate genes with mostly rare cytogenetic aberrations or single-gene mutations in 10–25% of cases . Copy number variants (CNVs) are reported in 44% of familial ASD cases and ~7–10% of sporadic cases[32]. However, only a small number of genes have been proven causative at this point and genetic testing results need to be reviewed critically in the context of the mutation, gene function, gene modifiers, and potential protein interaction networks, gene environment interactions, or sex-linked modifiers [33][34]. These causative genetic factors converge on mechanisms primarily related to neuronal development, plasticity, synaptic structure, and performance [35][36], e.g., neurexin (NRXN), neuroligin (NLGN), SH and multiple ankyrin repeat domains (SHANK), tuberous sclerosis 1/2 protein (TSC1/2), FMRP translational regulator 1 (FMR1), or methyl-CpG-binding protein 2 (MECP2), which are related to synaptic performance[35]. In addition, environmental factors have a significant contribution to disease risk of ASD as established in twins [37] and include prenatal viral infection, zinc deficiency, abnormal melatonin synthesis, maternal diabetes, parental age, and other potential postnatal risk and stress factors [38].
2. Overlapping Clinical Motor and Behavioral Phenomenology between ASD and PD
Loss of dopaminergic signaling in the nigro-striatal pathway is the primary cause for the motor symptoms in PD, causing an imbalance in the neuronal circuits of basal ganglia. In addition, it is becoming increasingly clear that the balance of the basal ganglia circuit is also altered in ASD, affecting both cognition and motor function [39][40]. Furthermore, it has been postulated that impairment in cortical control of striatal neuronal circuits underlies impulsive and compulsive activity—symptoms in both PD and ASD [41].
There are overlapping clinical phenomena in the cognitive and behavioral profiles of ASD and PD that can present independently of secondary causes, such as medication, e.g., neuroleptics or dopamine agonists [42]. In a clinical research study in two cohorts of ASD with an age over 39 years, there was a surprising increase in Parkinsonian motor features in 20% of cases (without neuroleptics use), and 7% of participants in this cohort were also clinically diagnosed with PD prior to the study [43]. Non-motor features are also commonly shared between ASD and PD, demonstrated by a higher prevalence of depression and anxiety, and obsessive–compulsive disorders (OCDs) [44]. Patients with ASD show a prevalence of anxiety in 39.6% [45], depression in ~30% [46][47][48][49], and OCD in 17.4% of cases. In PD, the prevalence of anxiety is described in 65–68.42% [50][51], depression in 13.8%–56% [50][51][52], OCD in 37.5% (drug naïve) [53], and impulse control behavior (ICB) in 17.1–28.6% [54][55] of patients with PD. ICB in PD is illustrated by gambling, compulsive eating, punding, excessive shopping, or hypersexuality. A recent study showed an improvement of ICB symptoms after changing medication from dopamine agonists to levodopa/carbidopa slow-release formulations [56].
There are also rare cases of co-morbidity of ASD and early onset PD. One clinical case report describes a patient, diagnosed with PD at the age of 30, with left-side-predominant hypokinetic-rigid syndrome. By history, the patient had delayed language and motor development. During childhood, he had problems with social contact and showing emotions. In a formal assessment for ASD, the patient scored positive on all three domains (deficits in social interaction, communication, and stereotypical repetitive behavior), but genetic analysis was not performed [57].
A study in 39 children with ASD (age 2–8 years) without known karyotypic abnormalities showed a significant 3-fold decrease in α-syn plasma levels versus controls, which links one of the key proteins in PD pathogenesis to ASD. In this study, beta-synuclein (β-syn) levels were also found to be significantly higher in ASD children compared to the control group, postulating a compensatory effect of β-syn [58]. This overlapping clinical phenomenology of motor and non-motor symptoms and the molecular link between ASD and PD point towards potentially converging disease mechanisms and imbalances of dopamine signaling in the basal ganglia.
In summary, we describe striking overlapping clinical symptoms of Parkinsonian motor problems in ASD and psychiatric symptoms, such as depression, anxiety, and impulsive and compulsive activity, in ASD and PD. Besides genetic causes, it will be important to assess the overlapping molecular pathology of ASD and PD, including the inflammatory response and mitochondrial dysfunction, that might even serve as early molecular biomarkers. In clinical practice, the commonality of these two diseases points towards the importance of an interdisciplinary clinical care team comprising of movement disorder specialists and psychiatrists for the clinical care and treatment of ASD and PD. This would allow not only for patients to be served better but also for insight to be gained into the common disease etiology and advancing the development of novel molecular treatments.
3. Genes Linked to Neurodegenerative Diseases Also Play a Role in Neurodevelopmental Disorders
Genetic linkage studies and GWAS have provided deep molecular insight into disease mechanisms for both PD and ASD. While in PD, point mutations and non-coding risk variants have been predominantly described, only CNV alterations have been described in ASD; however, in the next section, we describe the frequencies for CNVs in the PARK2 gene and 22q11.2 deletions, and FMR1 repeat expansions, which can present both with clinical symptoms of PD and ASD. However, α-syn CNVs play a major role and comprise about 50% of reported mutations . In the gene (SNCA), duplications and triplications have been identified as a rare cause of PD, although genomic SNCA deletions have not been described in the literature. The mining of public genomic databases, however, revealed cases heterozygous for genomic SNCA deletions, which also present with developmental delay and ASD [59] (Table S1).
3.1. PARK2 Copy Number Variants in PD and ASD and its Substrate GPR37 Are Linked to Autism
Point mutations and copy number variants in the PARK2 gene (PRKN, parkin RBR E3 ubiquitin protein ligase, OMIM *602544) have been implicated in autosomal recessive juvenile Parkinsonism first described in 1998 in Japanese families and is the most common genetic cause in early onset PD [60][61]. The PARK2 gene on chromosome 6q26 is the second-largest gene in the human genome and a common fragile site (FRA6E) in the human genome. PARK2 point mutations and CNVs have also been described in glioblastoma, ovarian, and liver cancers [62][63][64][65].
Strikingly, it has not been widely recognized that CNVs encompassing the PARK2 gene are also found in cohorts of patients with ASD/developmental delay and attention deficit hyperactivity disorder (ADHD) at a frequency of 0.5–2.2%. There are single clinical case reports of deletions and duplications of the PARK2 gene [66][67], but GWAS studies and clinical diagnostic case series also support these findings. The first whole-genome CNV study in 2009 detected PARK2 deletions in seven ASD cases (0.8%) and none in controls among 859 ASD cases and 1409 controls of European ancestry [68]. In a more recent CNV GWAS study in 335 ASD cases and 1093 healthy controls in Han Chinese, 6 CNVs (1.8%) were detected in the PARK2 region in the ASD cohort (four deletions and two duplications in exons 2–7, with a CNV size between 50 and 250kb), while only two duplications were found in controls (0.2%) [69]. Furthermore, in an ADHD cohort of 875 young patients and 2066 matched controls (test and replication groups combined), 16 PARK2 CNVs (1.8%) were identified in the ADHD cohort and 3 (0.1%) in the controls [70]. A diagnostic case series from Czech Republic describes two CNVs (2.2%) in the PARK2 gene, one deletion and one duplication out of 92 ASD cases [71]. A second consecutive diagnostic case series from the University Kansas included 215 patients referred for autism/ASD or developmental delay/learning disability and found one PARK2 CNV duplication (0.5%) in a patient with learning disability and dysmorphic features [72].
In the Autism Genome Project (AGP) cohort (European and Northern American ancestry), 15 out of 2446 ASD cases (0.61%) harbored partial exonic PARK2 CNV deletions or duplications [73] (Figure 1A, Table S1). We excluded 31 CNVs in this region that did not encompass exons and, hence, we could not establish causality according to the American College of Medical Genetics (ACMG) guidelines [74]. Of the 15 PARK2 CNVs included, 6 were deletions and 9 were duplications, spanning coding exons similar to CNV mutations found in juvenile PD .
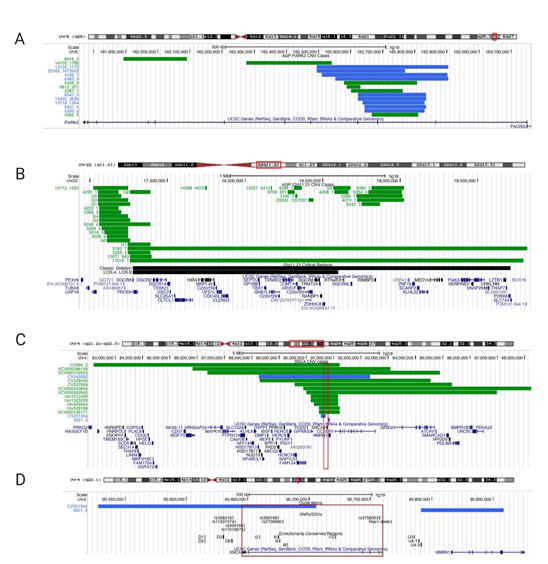
Figure 1. University of California Santa Cruz (UCSC) Genome browser view of copy number variants CNVs in PARK2, 22q11.2 deletion, and SNCA/4q22.1. (A) Partial exonic PARK2 CNV deletion/duplication coordinates for UCSC genome browser custom tracks (chr6q26; genome build GRCh37/hg19, February 2009); green bars depict deletions, blue bars depict duplications; case identifiers listed on the left) on chromosome 6q26 found in the Autism Genome Project (AGP) cohort. Table S1 lists genomic positions to build custom tracks in UCSC Genome Browser. (B) 22q11.21 deletions’ coordinates for UCSC genome browser custom tracks (genome build NCBI36/hg18, March 2006). As a reference in black bars, we included the 22q11.21 deletion region (Velocardiofacial/DiGeorge syndrome, https://decipher.sanger.ac.uk/syndrome/16#genotype/cnv/21/browser) and the recently characterized critical region for a higher rate of autism (LCR-A to LCR-B) [75] Case data were analyzed from AGP for chromosome 22q.11.21. For cases with identical deletions, we grouped cases into groups G1 to G9 (column ID/Group ID). Every green bar depicts a deletion case with a numerical identifier; multiple cases are shown per line for space considerations. Table S2 lists genomic positions for custom tracks. (C) 4q22.1deletion cases with neurodevelopmental delay and/or ASD (<10MB; chr4:82,210,925-98,235,479); (D) Smaller SNCA duplication cases mapped to regulatory elements (SNPs/SSVs and evolutionary conserved regions) in the UCSC genome browser (chr.4:90,527,679-90,858,538; genome build GRCh37/hg19, February 2009). Every case has a numerical identifier, the red box indicates SNCA genomic region. Regulatory elements are listed in black in panel B (reviewed in ). Table S3 lists the genomic positions to build UCSC Genome Browser custom tracks.
The PARK2 gene encodes parkin, an E3 ubiquitin-protein ligase, which is recruited to damaged mitochondria to ubiquitylate outer membrane proteins to promote clearance through mitophagy and autophagic pathways [76]. Parkin was also been found to interact with pyruvate kinase isozymes M2 (PKM2), regulating the glycolysis pathway[77]. Parkin has a number of substrates, such as mitochondrial proteins and endosomal trafficking regulators recently described in a drosophila screen, including vacuolar protein sorting-associated protein 35 and 4 (VPS35, VPS4), arginine kinase (ArgK), and reductive dehalogenase anchoring protein (RdhB) [78][79]. Relevant to neurodegeneration, Parkin has been reported to interact with a glycosylated form of α-syn (a-Sp22) [80]; synphilin, an α-synuclein-interacting protein [81]; and synaptotagmin XI (SYT11), involved in regulation of the synaptic vesicle pool and release [82][83][84]. In addition, parkin ubiquitinates the parkin-associated endothelin-receptor-like receptor (Pael-R/GPR37) and promotes degradation of its insoluble form [85]. Pael-R/GPR37 interacts with the dopamine transporter (DAT, SLC6A3) and modulates DAT activity [86]. Pael-R knockout mice show an increase in DAT expression and resistance to methyl-phenyl-tetrahydropyridine (MPTP) exposure. Loss of parkin function leads to accumulation of Pael-R/GPR37, which is described in post-mortem brains of PARK2 cases. Pael-R/GPR37 is also found in the core of LBs and Lewy neurites [87]. During neuronal development, GPR37 is required for Wnt-dependent neurogenesis and functions in the maturation of the N-terminal bulky b-propellers of the Wnt co-receptor low-density lipoprotein receptor (LDLR)-related protein 6 (LRP6) [88].
To date, no mutations or risk variants in the Pael-R/GPR37 gene have been described in PD; however, several mutations in the GPR37 gene (chr7q31–33, AUTS1 region) have been described in ASD patients. A c.1585–1587del TTC (Del312F) in a patient of Japanese descent, c.2324G > A (R558Q) in one Caucasian patient, and T589M was found in seven affected Caucasian patients [89]. Causative GPR37 mutations could lead to a loss of function, resulting in an increase of DAT expression and activity.
3.2. 22q11.2 Deletion/Di George Syndrome Linked to Parkinsonism
While PARK2 is a bona fide PD gene, deletions of chromosome 22q11.2 (22q11.2DS, OMIM # 188400) have only recently been implicated in PD [90]. The 22q11.2 deletion is considered to be one of the most common deletion syndromes (~1:1000–3000 births) and was first described in 1968 [91][92]. The majority of 22q11.2DS encompass a 3-Mb region including ~90 genes, whereas 5–10% of patients have a smaller 1.5-Mb deletion. About half of these genes are expressed in the human brain [93]. The 22q11.2 deletion is a heterogeneous clinical syndrome involving neurodevelopmental, psychiatric, cardiac, immunological, and endocrinological abnormalities. Children with 22q11.2DS have developmental delay and are at a higher risk for ASD, ADHD, and other mental disorders, including depression and anxiety [94]. The complex clinical presentation is due to the large number of genes in the region. Cases with smaller atypical CNVs in this region contribute to the understanding of the underlying causative genes, i.e., the critical region for a higher rate of autism encompasses the low copy repeat (LCR) region A to B and increases the risk of autism by ~40% (Figure 1B, Table S2). LCRs can mediate chromosomal rearrangements and there are a total of eight LCRs within 22q11 [95][96]. In the AGP project, 59 CNV deletion cases (ranging from 6.2 kb to 2.6 Mb) were detected within the 22q11.2 region in an ASD cohort (2.4%) (Figure 1B, Table S2). Ten cases had a deletion surrounding the LCR-A to LCR-B region . Several smaller deletions in the region could pinpoint putative candidate genes relevant in the context of ASD.
Recently, it has been recognized that adults with 22q11.2DS have a higher risk of developing PD [97] as illustrated by a PD prevalence of 5.9% in a cohort of 159 adults (ages 35–64 years) with 22q11.2DS [98]. Clinically, the onset of symptoms is asymmetric, with typical progressive motor features of bradykinesia, rigidity, and tremor. Patients respond well to levodopa therapy. PD onset in 22q11.2DS cases is often earlier, with a mean age 39.5 ± 8.5 years (range, 18–58) [99]. Other symptoms include early dystonia, a history of seizures, and neuropsychiatric symptoms (e.g., psychosis and anxiety) [100].
A mouse model encompassing the 22q11 genomic deletion region, the Df1/+ model [101], was found to have elevated levels of α-syn and p62, a key receptor for autophagy. They showed behavioral motor coordination deficits, which could be reversed by pharmacological inhibition of mammalian target of rapamycin (mTOR) activity by a rapamycin analog [102]. While it is difficult to pinpoint causative genes in this large deletion region implicated in PD or ASD, several genes would qualify as candidate genes., e.g., genes related to mitochondrial dysfunction, namely MRPL40, PRODH, SLC25A1, TANGO2, TXNRD2, and ZDDHC8. In this regard, disruption of a network of inner mitochondrial membrane transporters (SLC25A1-SLC25A4) required for synapse function has been identified in the Df1/+ mouse model and patient biomaterial from 22q11.2DS cases [103]. Another potential candidate gene is the catechol-O-methyltransferase (COMT) gene, which degrades catecholamines (i.e., dopamine or norepinephrine). Lower levels of COMT would lead to higher synaptic dopamine levels following neurotransmitter release, ultimately increasing dopaminergic stimulation at postsynaptic neurons and causing dopamine toxicity.
3.3. FMR1 Intermediate CGG Repeat Expansion Causes Autism and FXTAS/Parkinsonism
Variable CGG repeat expansions in the 5′ untranslated portion of the fragile mental retardation 1 gene (FMR1, chrXq27.3) cause a spectrum of disorders, including fragile X syndrome (FXS, OMIM # 300624) (> 200 CGG repeats), fragile X-associated tremor ataxia syndrome (FXTAS, OMIM # 300623; permutation 55–200 CGG repeats), and fragile X-associated primary ovarian insufficiency (FXPOI, premutation) [104].
Both the full repeat expansion and the premutation give rise to autism/ASD. It is estimated that 30% of FXS cases have autism and 2–6% of all cases with autism carry FMR1 repeat expansions. In addition, premutation carriers can develop FXTAS, a neurodegenerative disorder characterized by cerebellar ataxia, intention tremor, and cognitive impairment usually starting >65years of age. Interestingly, some patients with FMR1 premutations present with symptoms compatible with typical PD [105][106][107]. LB pathology has been described in FXTAS cases [108] but also tau pathology either consistent with Alzheimer’s disease or progressive supranuclear palsy (PSP) [109][110][111][112]. While the full FMR1 repeat expansion leads to deficiency or absence of the FMR1 protein (FMRP), the FMR1 premutation leads to RNA toxicity and results in eosinophilic intranuclear FMR1-mRNA-containing inclusions in neurons and astrocytes in various brain regions, including the cortex, basal ganglia, thalamus, hippocampus, amygdala, and SN [109]. The FMR1 protein functions as an RNA carrier protein controlling the translation of genes regulating synaptic development and plasticity.
3.4. SNCA Genomic Region (4q22.1) Is a Multiplication/Deletion Syndrome for PD and ASD
α-syn (encoded by SNCA, NM_000345.3, OMIM *163890) is a key protein in the pathogenesis of PD and related α-synucleinopathies [113]. Since its discovery as the first PD-causing gene in 1997 [114][115][116], several missense mutations, a number of risk SNPs, small structural variants (SSVs), and, recently, cases with SNCA multiplications and somatic mosaicism have been described [117][118]. Genomic duplications and triplications of various genomic sizes of the 4q22.1 genomic region are highly penetrant and contribute to PD, LBD, or multiple system atrophy (MSA). The critical genomic region of these cases spans the SNCA gene, which is the only gene overlapping in all described cases [118][119][120]. While earlier studies used single PCR-based copy number analysis, comparative genomic hybridization (CGH) and optical mapping can now define the size of CNVs, breakpoints, and orientation much more accurately [121] (Figure S1, Table S3C).
The underlying mechanism for these pathogenic duplications or triplications is intrachromosomal non-allelic homologous recombination (NAHR), which is a misalignment of homologous chromosomes during meiosis leading to the recombination and exchange of different regions of the chromosome, resulting in alterations of the genomic structure, such as duplications or deletions [122][123].
While multiplications of the SNCA locus are causative for neurodegeneration and LB pathology, deletions of this region on chromosome 4q22.1 have not been explicitly described as disease causing. In general, it is thought that lower levels of α-syn are possibly neuroprotective, hence, individuals with SNCA deletions would not contract disease. However, a closer look into public disease databases (e.g., ClinVar, Decipher, and ISCA) and large cohort studies for CNV variation in neurocognitive disorders reveals that there are patients with large deletions of the SNCA locus, which clinically present with developmental delay, ASD, and/or congenital abnormalities. Of note, these cases are distinct from chromosome 4q deletion syndromes, which encompass either a proximal deletion between 4(q11-q31) or distal deletion of 4(q31–q35) [124][125].
Fifteen SNCA CNV cases are described in the ClinVar and Decipher databases, ranging from 67 kb to 9 Mb, with the smallest overlapping region as the SNCA gene (Figure 1C and D, Table S3). These are cases submitted through clinical diagnostic labs and detailed clinical information is not available for these patients. In addition, there are several large cohort studies with children with developmental delay, intellectual disability, and/or ASD, in which cases with SNCA CNV deletions are described (Figure 1C and D, Table S3).
The Decipher CNV database lists three cases in total. Two of them have large deletions 5.75Mb (ID 337659, NCBI36/hg18; chr4:89571443-95328195) and 6.25Mb (ID 339430, NCBI36/hg18; chr4:88295927-94547516) with multiple genes affected, and a third case (ID 251304, NCBI36/hg18; chr 4:90527679-90705624) carrying a duplication of 177,946 bp partially spanning the SNCA gene only. This partial SNCA duplication is predicted to result in a loss of function of α-syn. The clinical presentation of this case is broadly described as having facial dysmorphology and intellectual disability. Another duplication was described in the AGP cohort upstream of the SNCA gene. While the pathogenicity is unclear, this CNV resides within a non-coding region of evolutionarily conserved elements that have been shown to modulate gene expression in cellular reporter assays (Figure 1D) [126].
Vulto-van Silfout et al. 2013 described a cohort of 5531 children, ages 3–17, with intellectual disabilities, ASD, and/or congenital abnormalities. They found CNVs in 25.1% of the cases using a 250,000 single-nucleotide polymorphism array platform. One patient carried an 884-kb deletion within the SNCA genomic region encompassing SNCA, MMRN1, and an unknown gene KIAA1680 (NCBI36/hg18; chr.4:90,629,298-91,514,095) [127]. Coe et al. 2014 described a large CNV analysis of 29,085 children with developmental delay and/or ASD in comparison to 19,584 healthy controls using array comparative genomic hybridization (CGH). Seven patients and two controls carried a 475-kb deletion (NCBI36/hg18; chr.4:90,793,560-91,268,616), including only the SNCA and MMRN1 genes [128].
3.5. Four PD-Associated Genes Are Linked with Neurodevelopmental Disorders
In summary, we have described genetic conditions that can give rise to ASD or PD/LBD depending on their gene dosage, establishing precedence that there are potential converging mechanisms and pathways that play a role both in neurodevelopment and neurodegenerative processes. Partial PARK2 deletions/duplications cause early onset PD when they lead to complete loss of protein in compound heterozygote or homozygote individuals, whereas heterozygote carriers are found in autism populations, which still express 50% of the parkin protein, but in these cases lead to haploinsufficiency. On the contrary, expansions of the FMR1 gene >200 CGG repeats lead to a loss of FMRP protein and autism, but the premutation causes PD, with the RNA found in foci in the brain of FXTAS cases . The 22q11 deletion can lead to both ASD and PD as a continuous symptom spectrum similar to cases with trisomy 21 and the development of Alzheimer’s disease [129[130]. Lastly, deletions and partial duplications of SNCA gene pinpoint to a new critical region for developmental delay and ASD on chromosome 4q22.1. We propose a new model for the SNCA multiplication/deletion region. While overexpression of the SNCA gene has been accepted as being causative for PD and related neurodegenerative conditions, we propose that deletions or partial duplications of the SNCA genomic regions lead to developmental delay and ASD and relevant models could shed light on this connection and mechanisms possible linked through synaptic function and neuronal development.
References
- Balestrino, R.; Schapira, A.H.V. Parkinson Disease. Eur. J. Neurol. 2019, 10.1111/ene.14108, doi:10.1111/ene.14108.
- Han, J.W.; Ahn, Y.D.; Kim, W.S.; Shin, C.M.; Jeong, S.J.; Song, Y.S.; Bae, Y.J.; Kim, J.M. Psychiatric Manifestation in Patients with Parkinson’s Disease. J. Korean Med. Sci. 2018, 33, e300, doi:10.3346/jkms.2018.33.e300.
- Ke, J.Q.; Shao, S.M.; Zheng, Y.Y.; Fu, F.W.; Zheng, G.Q.; Liu, C.F. Sympathetic skin response and heart rate variability in predicting autonomic disorders in patients with Parkinson disease. Medicine (Baltimore) 2017, 96, e6523, doi:10.1097/MD.0000000000006523.
- Moisan, F.; Kab, S.; Mohamed, F.; Canonico, M.; Le Guern, M.; Quintin, C.; Carcaillon, L.; Nicolau, J.; Duport, N.; Singh-Manoux, A.; et al. Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 952–957, doi:10.1136/jnnp-2015-312283.
- Langston, J.W.; Schüle, B.; Rees, L.; Nichols, R.J.; Barlow, C. Multisystem Lewy body disease and the other parkinsonian disorders. Nat. Genet. 2015, 47, 1378–1384, doi:10.1038/ng.3454.
- Ghebremedhin, E.; Del Tredici, K.; Langston, J.W.; Braak, H. Diminished tyrosine hydroxylase immunoreactivity in the cardiac conduction system and myocardium in Parkinson’s disease: An anatomical study. Acta. Neuropathol. 2009, 118, 777–784, doi:10.1007/s00401-009-0596-y.
- Kalia, L.V.; Kalia, S.K. alpha-Synuclein and Lewy pathology in Parkinson’s disease. Curr. Opin. Neurol. 2015, 28, 375–381, doi:10.1097/WCO.0000000000000215.
- Gomperts, S.N. Lewy Body Dementias: Dementia With Lewy Bodies and Parkinson Disease Dementia. Continuum (Minneap Minn) 2016, 22, 435–463, doi:10.1212/CON.0000000000000309.
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci.: Off. J. Soc. Neurosci. 2009, 29, 444–453, doi:10.1523/JNEUROSCI.4029-08.2009.
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol. 2009, 201, 1–119.
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454, doi:10.1038/nrneurol.2013.132.
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P. Environmental risk factors and Parkinson’s disease: An umbrella review of meta-analyses. Parkinsonism Relat. Disord. 2016, 23, 1–9, doi:10.1016/j.parkreldis.2015.12.008.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912, doi:10.1016/S0140-6736(14)61393-3.
- Delamarre, A.; Meissner, W.G. Epidemiology, environmental risk factors and genetics of Parkinson’s disease. Presse. Med. 2017, 46, 175–181, doi:10.1016/j.lpm.2017.01.001.
- Klemann, C.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Parkinsons Dis. 2017, 3, 14, doi:10.1038/s41531-017-0015-3.
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharm. Toxicol 2014, 54, 141–164, doi:10.1146/annurev-pharmtox-011613-135937.
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12, doi:10.1016/j.cger.2019.08.002.
- Klein, C.; Schneider, S.A.; Lang, A.E. Hereditary parkinsonism: Parkinson disease look-alikes--an algorithm for clinicians to “PARK” genes and beyond. Mov. Disord. 2009, 24, 2042–2058, doi:10.1002/mds.22675.
- Singleton, A.; Hardy, J. Progress in the genetic analysis of Parkinson’s disease. Hum. Mol. Genet. 2019, 28, R215-R218, doi:10.1093/hmg/ddz183.
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102, doi:10.1016/S1474-4422(19)30320-5.
- Tanner, C.M. Advances in environmental epidemiology. Mov. Disord. 2010, 25 Suppl 1, S58-62, doi:10.1002/mds.22721.
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An Overview of Autism Spectrum Disorder, Heterogeneity and Treatment Options. Neurosci. Bull 2017, 33, 183–193, doi:10.1007/s12264-017-0100-y.
- Emberti Gialloreti, L.; Curatolo, P. Autism Spectrum Disorder: Why Do We Know So Little? Front. Neurol. 2018, 9, 670, doi:10.3389/fneur.2018.00670.
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910, doi:10.1016/S0140-6736(13)61539-1.
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell Neurosci. 2018, 12, 470, doi:10.3389/fncel.2018.00470.
- Huguet, G.; Ey, E.; Bourgeron, T. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genom. Hum. Genet. 2013, 14, 191–213, doi:10.1146/annurev-genom-091212-153431.
- Lange, N.; Travers, B.G.; Bigler, E.D.; Prigge, M.B.; Froehlich, A.L.; Nielsen, J.A.; Cariello, A.N.; Zielinski, B.A.; Anderson, J.S.; Fletcher, P.T.; et al. Longitudinal volumetric brain changes in autism spectrum disorder ages 6-35 years. Autism Res. 2015, 8, 82–93, doi:10.1002/aur.1427.
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145, doi:10.1016/j.tins.2007.12.005.
- Allman, J.M.; Tetreault, N.A.; Hakeem, A.Y.; Manaye, K.F.; Semendeferi, K.; Erwin, J.M.; Park, S.; Goubert, V.; Hof, P.R. The von Economo neurons in the frontoinsular and anterior cingulate cortex. Ann. N Y Acad. Sci. 2011, 1225, 59–71, doi:10.1111/j.1749-6632.2011.06011.x.
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta. Neuropathol. 2017, 134, 537–566, doi:10.1007/s00401-017-1736-4.
- Ecker, C.; Schmeisser, M.J.; Loth, E.; Murphy, D.G. Neuroanatomy and Neuropathology of Autism Spectrum Disorder in Humans. Adv. Anat. Embryol. Cell Biol. 2017, 224, 27–48, doi:10.1007/978-3-319-52498-6_2.
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488, doi:10.1016/j.ajhg.2007.12.009.
- Shen, J.; Lincoln, S.; Miller, D.T. Advances in Genetic Discovery and Implications for Counseling of Patients and Families with Autism Spectrum Disorders. Curr. Genet. Med. Rep. 2014, 2, 124–134, doi:10.1007/s40142-014-0047-5.
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell Neurosci. 2019, 13, 385, doi:10.3389/fncel.2019.00385.
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect Biol. 2012, 4, doi:10.1101/cshperspect.a009886.
- Kleijer, K.T.E.; Huguet, G.; Tastet, J.; Bourgeron, T.; Burbach, J.P.H. Anatomy and Cell Biology of Autism Spectrum Disorder: Lessons from Human Genetics. Adv. Anat. Embryol. Cell Biol. 2017, 224, 1–25, doi:10.1007/978-3-319-52498-6_1.
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102, doi:10.1001/archgenpsychiatry.2011.76.
- Grabrucker, A.M. Environmental factors in autism. Front. Psychiatry 2012, 3, 118, doi:10.3389/fpsyt.2012.00118.
- Subramanian, K.; Brandenburg, C.; Orsati, F.; Soghomonian, J.J.; Hussman, J.P.; Blatt, G.J. Basal ganglia and autism—a translational perspective. Autism Res. 2017, 10, 1751–1775, doi:10.1002/aur.1837.
- Paval, D. A Dopamine Hypothesis of Autism Spectrum Disorder. Dev. Neurosci. 2017, 39, 355–360, doi:10.1159/000478725.
- Fineberg, N.A.; Potenza, M.N.; Chamberlain, S.R.; Berlin, H.A.; Menzies, L.; Bechara, A.; Sahakian, B.J.; Robbins, T.W.; Bullmore, E.T.; Hollander, E. Probing compulsive and impulsive behaviors, from animal models to endophenotypes: A narrative review. Neuropsychopharmacology 2010, 35, 591–604, doi:10.1038/npp.2009.185.
- Hollander, E.; Wang, A.T.; Braun, A.; Marsh, L. Neurological considerations: Autism and Parkinson’s disease. Psychiatry Res. 2009, 170, 43–51, doi:10.1016/j.psychres.2008.07.014.
- Starkstein, S.; Gellar, S.; Parlier, M.; Payne, L.; Piven, J. High rates of parkinsonism in adults with autism. J. Neurodev. Disord. 2015, 7, 29, doi:10.1186/s11689-015-9125-6.
- Strang, J.F.; Kenworthy, L.; Daniolos, P.; Case, L.; Wills, M.C.; Martin, A.; Wallace, G.L. Depression and Anxiety Symptoms in Children and Adolescents with Autism Spectrum Disorders without Intellectual Disability. Res. Autism Spectr. Disord. 2012, 6, 406–412, doi:10.1016/j.rasd.2011.06.015.
- Postorino, V.; Kerns, C.M.; Vivanti, G.; Bradshaw, J.; Siracusano, M.; Mazzone, L. Anxiety Disorders and Obsessive-Compulsive Disorder in Individuals with Autism Spectrum Disorder. Curr. Psychiatry Rep. 2017, 19, 92, doi:10.1007/s11920-017-0846-y.
- Wijnhoven, L.A.; Niels-Kessels, H.; Creemers, D.H.; Vermulst, A.A.; Otten, R.; Engels, R.C. Prevalence of comorbid depressive symptoms and suicidal ideation in children with autism spectrum disorder and elevated anxiety symptoms. J. Child Adolesc. Ment. Health 2019, 31, 77–84, doi:10.2989/17280583.2019.1608830.
- Pezzimenti, F.; Han, G.T.; Vasa, R.A.; Gotham, K. Depression in Youth with Autism Spectrum Disorder. Child Adolesc. Psychiatr. Clin. N. Am. 2019, 28, 397–409, doi:10.1016/j.chc.2019.02.009.
- Dell’Osso, L.; Carpita, B.; Muti, D.; Morelli, V.; Salarpi, G.; Salerni, A.; Scotto, J.; Massimetti, G.; Gesi, C.; Ballerio, M.; et al. Mood symptoms and suicidality across the autism spectrum. Compr. Psychiatry 2019, 91, 34–38, doi:10.1016/j.comppsych.2019.03.004.
- Rai, D.; Heuvelman, H.; Dalman, C.; Culpin, I.; Lundberg, M.; Carpenter, P.; Magnusson, C. Association Between Autism Spectrum Disorders With or Without Intellectual Disability and Depression in Young Adulthood. Jama. Netw. Open 2018, 1, e181465, doi:10.1001/jamanetworkopen.2018.1465.
- Hermanowicz, N.; Jones, S.A.; Hauser, R.A. Impact of non-motor symptoms in Parkinson’s disease: A PMDAlliance survey. Neuropsychiatr. Dis. Treat. 2019, 15, 2205–2212, doi:10.2147/NDT.S213917.
- Chuquilin-Arista, F.; Alvarez-Avellon, T.; Menendez-Gonzalez, M. Prevalence of Depression and Anxiety in Parkinson Disease and Impact on Quality of Life: A Community-Based Study in Spain. J. Geriatr. Psychiatry Neurol. 2019, 10.1177/0891988719874130, 891988719874130, doi:10.1177/0891988719874130.
- Poletti, M.; Lucetti, C.; Del Dotto, P.; Berti, C.; Logi, C.; Bonuccelli, U. Relationship between neuropsychiatric symptoms and cognitive performance in de novo Parkinson’s disease. J. Neuropsychiatry Clin. Neurosci. 2012, 24, E22-23, doi:10.1176/appi.neuropsych.11100243.
- Nicoletti, A.; Luca, A.; Raciti, L.; Contrafatto, D.; Bruno, E.; Dibilio, V.; Sciacca, G.; Mostile, G.; Petralia, A.; Zappia, M. Obsessive compulsive personality disorder and Parkinson’s disease. PLoS ONE 2013, 8, e54822, doi:10.1371/journal.pone.0054822.
- Antonini, A.; Barone, P.; Bonuccelli, U.; Annoni, K.; Asgharnejad, M.; Stanzione, P. ICARUS study: Prevalence and clinical features of impulse control disorders in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 317–324, doi:10.1136/jnnp-2016-315277.
- Gatto, E.M.; Aldinio, V. Impulse Control Disorders in Parkinson’s Disease. A Brief and Comprehensive Review. Front. Neurol. 2019, 10, 351, doi:10.3389/fneur.2019.00351.
- Lee, J.Y.; Jeon, B.; Koh, S.B.; Yoon, W.T.; Lee, H.W.; Kwon, O.D.; Kim, J.W.; Kim, J.M.; Ma, H.I.; Kim, H.T.; et al. Behavioural and trait changes in parkinsonian patients with impulse control disorder after switching from dopamine agonist to levodopa therapy: Results of REIN-PD trial. J Neurol. Neurosurg. Psychiatry 2019, 90, 30–37, doi:10.1136/jnnp-2018-318942.
- Oudkerk, M.; Esselink, R.A.J.; Kan, C.C.; Tendolkar, I.; van Beek, M.H.C.T. Impact of Comorbid Autism Spectrum Disorder in an Individual with Idiopathic Young-Onset Parkinson’s Disease. Adv. Neurodev. Disord. 2019, 3, 91–94, doi:10.1007/s41252-018-0089-x.
- Sriwimol, W.; Limprasert, P. Significant Changes in Plasma Alpha-Synuclein and Beta-Synuclein Levels in Male Children with Autism Spectrum Disorder. Biomed. Res. Int. 2018, 2018, 4503871, doi:10.1155/2018/4503871.
- Piper, D.A.; Sastre, D.; Schüle, B. Advancing Stem Cell Models of Alpha-Synuclein Gene Regulation in Neurodegenerative Disease. Front. Neurosci. 2018, 12, 199, doi:10.3389/fnins.2018.00199.
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608, doi:10.1038/33416.
- Hedrich, K.; Eskelson, C.; Wilmot, B.; Marder, K.; Harris, J.; Garrels, J.; Meija-Santana, H.; Vieregge, P.; Jacobs, H.; Bressman, S.B.; et al. Distribution, type, and origin of Parkin mutations: Review and case studies. Mov. Disord. 2004, 19, 1146–1157, doi:10.1002/mds.20234.
- Denison, S.R.; Wang, F.; Becker, N.A.; Schüle, B.; Kock, N.; Phillips, L.A.; Klein, C.; Smith, D.I. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 2003, 22, 8370–8378, doi:10.1038/sj.onc.1207072.
- Schüle, B.; Byrne, C.; Rees, L.; Langston, J.W. Is PARKIN parkinsonism a cancer predisposition syndrome? Neurol. Genet. 2015, 1, e31, doi:10.1212/NXG.0000000000000031.
- Wang, F.; Denison, S.; Lai, J.P.; Philips, L.A.; Montoya, D.; Kock, N.; Schüle, B.; Klein, C.; Shridhar, V.; Roberts, L.R.; et al. Parkin gene alterations in hepatocellular carcinoma. Genes Chromosomes Cancer 2004, 40, 85–96, doi:10.1002/gcc.20020.
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 2010, 42, 77–82, doi:10.1038/ng.491.
- Scheuerle, A.; Wilson, K. PARK2 copy number aberrations in two children presenting with autism spectrum disorder: Further support of an association and possible evidence for a new microdeletion/microduplication syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156B, 413–420, doi:10.1002/ajmg.b.31176.
- Mariani, M.; Crosti, F.; Redaelli, S.; Fossati, C.; Piras, R.; Biondi, A.; Dalpra, L.; Selicorni, A. Partial duplication of the PARK2 gene in a child with developmental delay and her normal mother: A second report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162B, 485–486, doi:10.1002/ajmg.b.32173.
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573, doi:10.1038/nature07953.
- Yin, C.L.; Chen, H.I.; Li, L.H.; Chien, Y.L.; Liao, H.M.; Chou, M.C.; Chou, W.J.; Tsai, W.C.; Chiu, Y.N.; Wu, Y.Y.; et al. Genome-wide analysis of copy number variations identifies PARK2 as a candidate gene for autism spectrum disorder. Mol. Autism 2016, 7, 23, doi:10.1186/s13229-016-0087-7.
- Jarick, I.; Volckmar, A.L.; Putter, C.; Pechlivanis, S.; Nguyen, T.T.; Dauvermann, M.R.; Beck, S.; Albayrak, O.; Scherag, S.; Gilsbach, S.; et al. Genome-wide analysis of rare copy number variations reveals PARK2 as a candidate gene for attention-deficit/hyperactivity disorder. Mol. Psychiatry 2014, 19, 115–121, doi:10.1038/mp.2012.161.
- Capkova, P.; Srovnal, J.; Capkova, Z.; Staffova, K.; Becvarova, V.; Trkova, M.; Adamova, K.; Santava, A.; Curtisova, V.; Hajduch, M.; et al. MLPA is a practical and complementary alternative to CMA for diagnostic testing in patients with autism spectrum disorders and identifying new candidate CNVs associated with autism. PeerJ 2019, 6, e6183, doi:10.7717/peerj.6183.
- Roberts, J.L.; Hovanes, K.; Dasouki, M.; Manzardo, A.M.; Butler, M.G. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene 2014, 535, 70–78, doi:10.1016/j.gene.2013.10.020.
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L. Clinical significance of de novo and inherited copy-number variation; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694, doi:10.1016/j.ajhg.2014.03.018.
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 2019, 10.1038/s41436-019-0655-2, doi:10.1038/s41436-019-0655-2.
- Clements, C.C.; Wenger, T.L.; Zoltowski, A.R.; Bertollo, J.R.; Miller, J.S.; de Marchena, A.B.; Mitteer, L.M.; Carey, J.C.; Yerys, B.E.; Zackai, E.H.; et al. Critical region within 22q11.2 linked to higher rate of autism spectrum disorder. Mol. Autism 2017, 8, 58, doi:10.1186/s13229-017-0171-7.
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999, doi:10.1101/gad.262758.115.
- Liu, K.; Li, F.; Han, H.; Chen, Y.; Mao, Z.; Luo, J.; Zhao, Y.; Zheng, B.; Gu, W.; Zhao, W. Parkin Regulates the Activity of Pyruvate Kinase M2. J. Biol. Chem. 2016, 291, 10307–10317, doi:10.1074/jbc.M115.703066.
- Martinez, A.; Lectez, B.; Ramirez, J.; Popp, O.; Sutherland, J.D.; Urbe, S.; Dittmar, G.; Clague, M.J.; Mayor, U. Quantitative proteomic analysis of Parkin substrates in Drosophila neurons. Mol. Neurodegener 2017, 12, 29, doi:10.1186/s13024-017-0170-3.
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 2015, 7, 248, doi:10.3389/fnagi.2015.00248.
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269, doi:10.1126/science.1060627.
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: Implications for Lewy-body formation in Parkinson disease. Nat. Med. 2001, 7, 1144–1150, doi:10.1038/nm1001-1144.
- Huynh, D.P.; Scoles, D.R.; Nguyen, D.; Pulst, S.M. The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum. Mol. Genet. 2003, 12, 2587–2597, doi:10.1093/hmg/ddg269.
- Wang, C.; Kang, X.; Zhou, L.; Chai, Z.; Wu, Q.; Huang, R.; Xu, H.; Hu, M.; Sun, X.; Sun, S.; et al. Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson’s disease-like pathology. Nat. Commun. 2018, 9, 81, doi:10.1038/s41467-017-02593-y.
- Shimojo, M.; Madara, J.; Pankow, S.; Liu, X.; Yates, J., 3rd; Sudhof, T.C.; Maximov, A. Synaptotagmin-11 mediates a vesicle trafficking pathway that is essential for development and synaptic plasticity. Genes Dev. 2019, 33, 365–376, doi:10.1101/gad.320077.118.
- Leinartaite, L.; Svenningsson, P. Folding Underlies Bidirectional Role of GPR37/Pael-R in Parkinson Disease. Trends Pharm. Sci. 2017, 38, 749–760, doi:10.1016/j.tips.2017.05.006.
- Marazziti, D.; Mandillo, S.; Di Pietro, C.; Golini, E.; Matteoni, R.; Tocchini-Valentini, G.P. GPR37 associates with the dopamine transporter to modulate dopamine uptake and behavioral responses to dopaminergic drugs. Proc. Natl. Acad. Sci. USA 2007, 104, 9846–9851, doi:10.1073/pnas.0703368104.
- Murakami, T.; Shoji, M.; Imai, Y.; Inoue, H.; Kawarabayashi, T.; Matsubara, E.; Harigaya, Y.; Sasaki, A.; Takahashi, R.; Abe, K. Pael-R is accumulated in Lewy bodies of Parkinson’s disease. Ann. Neurol. 2004, 55, 439–442, doi:10.1002/ana.20064.
- Berger, B.S.; Acebron, S.P.; Herbst, J.; Koch, S.; Niehrs, C. Parkinson’s disease-associated receptor GPR37 is an ER chaperone for LRP6. Embo. Rep. 2017, 18, 712–725, doi:10.15252/embr.201643585.
- Fujita-Jimbo, E.; Yu, Z.L.; Li, H.; Yamagata, T.; Mori, M.; Momoi, T.; Momoi, M.Y. Mutation in Parkinson disease-associated, G-protein-coupled receptor 37 (GPR37/PaelR) is related to autism spectrum disorder. PLoS ONE 2012, 7, e51155, doi:10.1371/journal.pone.0051155.
- Fung, W.; Peall, K.J. Does 22q11.2 deletion syndrome contribute to the genetic aetiology of Parkinson’s disease? J. Neurol. 2018, 265, 2463–2465, doi:10.1007/s00415-018-9046-x.
- Kobrynski, L.J.; Sullivan, K.E. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes. Lancet 2007, 370, 1443–1452, doi:10.1016/S0140-6736(07)61601-8.
- Kirkpatrick, J.A., Jr.; DiGeorge, A.M. Congenital absence of the thymus. Am. J. Roentgenol. Radium Nucl. Med. 1968, 103, 32–37, doi:10.2214/ajr.103.1.32.
- Guna, A.; Butcher, N.J.; Bassett, A.S. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J Neurodev. Disord. 2015, 7, 18, doi:10.1186/s11689-015-9113-x.
- Schneider, M.; Debbane, M.; Bassett, A.S.; Chow, E.W.; Fung, W.L.; van den Bree, M.; Owen, M.; Murphy, K.C.; Niarchou, M.; Kates, W.R.; et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: Results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry 2014, 171, 627–639, doi:10.1176/appi.ajp.2013.13070864.
- Shaikh, T.H.; Kurahashi, H.; Saitta, S.C.; O’Hare, A.M.; Hu, P.; Roe, B.A.; Driscoll, D.A.; McDonald-McGinn, D.M.; Zackai, E.H.; Budarf, M.L.; et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000, 9, 489–501, doi:10.1093/hmg/9.4.489.
- Demaerel, W.; Mostovoy, Y.; Yilmaz, F.; Vervoort, L.; Pastor, S.; Hestand, M.S.; Swillen, A.; Vergaelen, E.; Geiger, E.A.; Coughlin, C.R.; et al. The 22q11 low copy repeats are characterized by unprecedented size and structural variability. Genome Res. 2019, 29, 1389–1401, doi:10.1101/gr.248682.119.
- Boot, E.; Bassett, A.S.; Marras, C. 22q11.2 Deletion Syndrome-Associated Parkinson’s Disease. Mov. Disord. Clin. Pr. 2019, 6, 11–16, doi:10.1002/mdc3.12687.
- Butcher, N.J.; Marras, C.; Pondal, M.; Rusjan, P.; Boot, E.; Christopher, L.; Repetto, G.M.; Fritsch, R.; Chow, E.W.C.; Masellis, M.; et al. Neuroimaging and clinical features in adults with a 22q11.2 deletion at risk of Parkinson’s disease. Brain 2017, 140, 1371–1383, doi:10.1093/brain/awx053.
- Boot, E.; Butcher, N.J.; Udow, S.; Marras, C.; Mok, K.Y.; Kaneko, S.; Barrett, M.J.; Prontera, P.; Berman, B.D.; Masellis, M.; et al. Typical features of Parkinson disease and diagnostic challenges with microdeletion 22q11.2. Neurology 2018, 90, e2059–e2067, doi:10.1212/WNL.0000000000005660.
- Boot, E.; Butcher, N.J.; van Amelsvoort, T.A.; Lang, A.E.; Marras, C.; Pondal, M.; Andrade, D.M.; Fung, W.L.; Bassett, A.S. Movement disorders and other motor abnormalities in adults with 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2015, 167A, 639–645, doi:10.1002/ajmg.a.36928.
- Lindsay, E.A.; Botta, A.; Jurecic, V.; Carattini-Rivera, S.; Cheah, Y.C.; Rosenblatt, H.M.; Bradley, A.; Baldini, A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 1999, 401, 379–383, doi:10.1038/43900.
- Sumitomo, A.; Horike, K.; Hirai, K.; Butcher, N.; Boot, E.; Sakurai, T.; Nucifora, F.C., Jr.; Bassett, A.S.; Sawa, A.; Tomoda, T. A mouse model of 22q11.2 deletions: Molecular and behavioral signatures of Parkinson’s disease and schizophrenia. Sci. Adv. 2018, 4, eaar6637, doi:10.1126/sciadv.aar6637.
- Gokhale, A.; Hartwig, C.; Freeman, A.A.H.; Bassell, J.L.; Zlatic, S.A.; Sapp Savas, C.; Vadlamudi, T.; Abudulai, F.; Pham, T.T.; Crocker, A.; et al. Systems Analysis of the 22q11.2 Microdeletion Syndrome Converges on a Mitochondrial Interactome Necessary for Synapse Function and Behavior. J. Neurosci.: Off. J. Soc. Neurosci. 2019, 39, 3561–3581, doi:10.1523/JNEUROSCI.1983-18.2019.
- Hagerman, R.; Hoem, G.; Hagerman, P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol. Autism 2010, 1, 12, doi:10.1186/2040-2392-1-12.
- Hall, D.A.; Berry-Kravis, E.; Zhang, W.; Tassone, F.; Spector, E.; Zerbe, G.; Hagerman, P.J.; Ouyang, B.; Leehey, M.A. FMR1 gray-zone alleles: Association with Parkinson’s disease in women? Mov. Disord. 2011, 26, 1900–1906, doi:10.1002/mds.23755.
- Hall, D.A.; Howard, K.; Hagerman, R.; Leehey, M.A. Parkinsonism in FMR1 premutation carriers may be indistinguishable from Parkinson disease. Parkinsonism Relat. Disord. 2009, 15, 156–159, doi:10.1016/j.parkreldis.2008.04.037.
- Niu, Y.Q.; Yang, J.C.; Hall, D.A.; Leehey, M.A.; Tassone, F.; Olichney, J.M.; Hagerman, R.J.; Zhang, L. Parkinsonism in fragile X-associated tremor/ataxia syndrome (FXTAS): Revisited. Parkinsonism Relat. Disord. 2014, 20, 456–459, doi:10.1016/j.parkreldis.2014.01.006.
- De Pablo-Fernandez, E.; Doherty, K.M.; Holton, J.L.; Revesz, T.; Djamshidian, A.; Limousin, P.; Bhatia, K.P.; Warner, T.T.; Lees, A.J.; Ling, H. Concomitant fragile X-associated tremor ataxia syndrome and Parkinson’s disease: A clinicopathological report of two cases. J. Neurol. Neurosurg. Psychiatry 2015, 86, 934–936, doi:10.1136/jnnp-2014-309460.
- Krans, A.; Skariah, G.; Zhang, Y.; Bayly, B.; Todd, P.K. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta. Neuropathol. Commun. 2019, 7, 152, doi:10.1186/s40478-019-0782-7.
- Sacino, A.N.; Prokop, S.; Walsh, M.A.; Adamson, J.; Subramony, S.H.; Krans, A.; Todd, P.K.; Giasson, B.I.; Yachnis, A.T. Fragile X-associated tremor ataxia syndrome with co-occurrent progressive supranuclear palsy-like neuropathology. Acta. Neuropathol. Commun. 2019, 7, 158, doi:10.1186/s40478-019-0818-z.
- Paucar, M.; Nennesmo, I.; Svenningsson, P. Pathological Study of a FMR1 Premutation Carrier With Progressive Supranuclear Palsy. Front. Genet. 2018, 9, 317, doi:10.3389/fgene.2018.00317.
- Tassone, F.; Greco, C.M.; Hunsaker, M.R.; Seritan, A.L.; Berman, R.F.; Gane, L.W.; Jacquemont, S.; Basuta, K.; Jin, L.W.; Hagerman, P.J.; et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav. 2012, 11, 577–585, doi:10.1111/j.1601-183X.2012.00779.x.
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernandez, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Arguero-Sanchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-Synuclein Physiology and Pathology: A Perspective on Cellular Structures and Organelles. Front. Neurosci. 2019, 13, 1399, doi:10.3389/fnins.2019.01399.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Nussbaum, R.L.; Polymeropoulos, M.H. Genetics of Parkinson’s disease. Hum. Mol. Genet. 1997, 6, 1687–1691.
- Munoz, E.; Oliva, R.; Obach, V.; Marti, M.J.; Pastor, P.; Ballesta, F.; Tolosa, E. Identification of Spanish familial Parkinson’s disease and screening for the Ala53Thr mutation of the alpha-synuclein gene in early onset patients. Neurosci. Lett. 1997, 235, 57–60.
- Mokretar, K.; Pease, D.; Taanman, J.W.; Soenmez, A.; Ejaz, A.; Lashley, T.; Ling, H.; Gentleman, S.; Houlden, H.; Holton, J.L.; et al. Somatic copy number gains of alpha-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 2018, 141, 2419–2431, doi:10.1093/brain/awy157.
- Fuchs, J.; Nilsson, C.; Kachergus, J.; Munz, M.; Larsson, E.M.; Schüle, B.; Langston, J.W.; Middleton, F.A.; Ross, O.A.; Hulihan, M.; et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 2007, 68, 916–922, doi:10.1212/01.wnl.0000254458.17630.c5.
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 2008, 63, 743–750, doi:10.1002/ana.21380.
- Book, A.; Guella, I.; Candido, T.; Brice, A.; Hattori, N.; Jeon, B.; Farrer, M.J.; Consortium, S.M.I.o.t.G. A Meta-Analysis of alpha-Synuclein Multiplication in Familial Parkinsonism. Front. Neurol. 2018, 9, 1021, doi:10.3389/fneur.2018.01021.
- Zafar, F.; Valappil, R.A.; Kim, S.; Johansen, K.K.; Chang, A.L.S.; Tetrud, J.W.; Eis, P.S.; Hatchwell, E.; Langston, J.W.; Dickson, D.W.; et al. Genetic fine-mapping of the Iowan SNCA gene triplication in a patient with Parkinson’s disease. NPJ Parkinsons Dis. 2018, 4, 18, doi:10.1038/s41531-018-0054-4.
- Parks, M.M.; Lawrence, C.E.; Raphael, B.J. Detecting non-allelic homologous recombination from high-throughput sequencing data. Genome Biol. 2015, 16, 72, doi:10.1186/s13059-015-0633-1.
- Liu, P.; Lacaria, M.; Zhang, F.; Withers, M.; Hastings, P.J.; Lupski, J.R. Frequency of nonallelic homologous recombination is correlated with length of h Genetic fine-mapping of the Iowan SNCA gene triplication in a patient with Parkinson's disease omology: Evidence that ectopic synapsis precedes ectopic crossing-over. Am. J. Hum. Genet. 2011, 89, 580–588, doi:10.1016/j.ajhg.2011.09.009.
- Vona, B.; Nanda, I.; Neuner, C.; Schroder, J.; Kalscheuer, V.M.; Shehata-Dieler, W.; Haaf, T. Terminal chromosome 4q deletion syndrome in an infant with hearing impairment and moderate syndromic features: Review of literature. BMC Med. Genet. 2014, 15, 72, doi:10.1186/1471-2350-15-72.
- Lin, A.E.; Garver, K.L.; Diggans, G.; Clemens, M.; Wenger, S.L.; Steele, M.W.; Jones, M.C.; Israel, J. Interstitial and terminal deletions of the long arm of chromosome 4: Further delineation of phenotypes. Am. J. Med. Genet. 1988, 31, 533–548, doi:10.1002/ajmg.1320310308.
- Sterling, L.; Walter, M.; Ting, D.; Schüle, B. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. F1000Res 2014, 3, 259, doi:10.12688/f1000research.3281.2.
- Vulto-van Silfhout, A.T.; Hehir-Kwa, J.Y.; van Bon, B.W.; Schuurs-Hoeijmakers, J.H.; Meader, S.; Hellebrekers, C.J.; Thoonen, I.J.; de Brouwer, A.P.; Brunner, H.G.; Webber, C.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Hum. Mutat. 2013, 34, 1679–1687, doi:10.1002/humu.22442.
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A.; van Bon, B.W.; Vulto-van Silfhout, A.T.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.E.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071, doi:10.1038/ng.3092.
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147, doi:10.1038/s41582-018-0132-6.
- Head, E.; Powell, D.; Gold, B.T.; Schmitt, F.A. Alzheimer’s Disease in Down Syndrome. Eur. J. Neurodegener. Dis. 2012, 1, 353–364.

