 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Suxiang Chen | + 2341 word(s) | 2341 | 2021-11-10 10:58:42 | | | |
| 2 | Vicky Zhou | Meta information modification | 2341 | 2021-11-25 01:40:14 | | |
Video Upload Options
Antisense oligonucleotides (AOs) have been developed to inhibit the production of alternatively spliced carcinogenic isoforms through splice modulation or mRNA degradation. AOs can also be used to induce splice switching, where the expression of an oncogenic protein can be inhibited by the induction of a premature stop codon. In general, AOs are modified chemically to increase their stability and binding affinity. One of the major concerns with AOs is efficient delivery. Strategies for the delivery of AOs are constantly being evolved to facilitate the entry of AOs into cells.
1. Introduction
RNA splicing is a form of RNA processing in which a freshly made precursor messenger RNA (pre-mRNA, or primary transcript) is transformed into a mature mRNA. The process of splicing is catalyzed by the RNA-protein complex known as the spliceosome. The spliceosome consists of five small nuclear ribonucleoproteins (snRNPs), U1, U2, U4, U5 and U6 [1]. During splicing, introns are removed, and exons are joined together.
Splicing can create a range of transcript variants by varying the exon composition of the same transcript. This phenomenon is termed alternative splicing. Alternative splicing is an indispensable process for expanding the spatiotemporal complexity of the transcriptome [2]. More specifically, it is the process wherein the exons of any primary transcript get rearranged to form an mRNA splice variant, and subsequently, the protein isoform, which are distinct both structurally and functionally [3]. In humans, more than 70% of multiexon genes undergo alternative splicing [4]. These splicing events are very tightly regulated and are highly tissue-specific [5]. Different cis- and trans-acting factors regulate the splicing process as they determine the splice site selection [6]. These splice sites can be weakened or strengthened due to point mutations, ultimately changing the splicing events, resulting in disease [6]. Furthermore, in most eukaryotic organisms, there are certain sequences that are ignored by the splicing machinery. These are known as the pseudoexons. In recent years, it has been reported that atypical inclusion of pseudoexons is more frequent than previously understood and is implicated in many human diseases, such as Duchenne muscular dystrophy (DMD) [7].
Cancer is caused by abnormal cell proliferation resulting from altered gene expression [8]. This can include overexpression of oncogenes [9] and production of aberrant splice variants, which subsequently leads to synthesis of carcinogenic protein isoforms [10]. In the past few decades, new developments into understanding the biological basis for cancer progression and therapeutic resistance have led to the identification of many alternative splicing events, which are key in the development of cancer. These carcinogenic splice isoforms are involved in various cellular processes, such as apoptosis, cell signaling and proliferation [11].
Strategies to inhibit the expression of oncogenes and the carcinogenic splice variants of essential genes at the mRNA level have become popular in the past few decades. Pre-mRNA or mRNA with known nucleotide sequences offer a chance to design antisense oligonucleotides (AOs) specific to the transcripts. AOs are short 15–30-base long DNA or RNA molecules that are chemically modified [12]. AOs are designed to be complementary to the target sequence. AOs bind to the target RNA through Watson–Crick base pairing. The specificity of AOs depends on the accuracy of the Watson–Crick base pairing on the estimate that a particular sequence of >16 bases occurs only once in the human genome [13]. Typically, AOs are chemically modified to improve target binding affinity and stability [14][15]. Efficient delivery of AOs into target cells is challenging. Strategies for the delivery of AOs are continually improved to ease the entry of the AOs into the cells [16].
Mechanisms of action of AOs are elucidated in the next section. AOs that induce RNase H-mediated mRNA degradation have been widely used for downregulation of oncogenes [11][17][18]; therefore, this review focuses primarily on six distinct studies that use AO-mediated splice modulation as a therapeutic strategy, with a discussion of different chemical modifications used and delivery strategies employed. In addition, although premature termination of a codon induced by AO-mediated exon skipping has not yet been reported for the development of anticancer AOs, this can potentially be a useful strategy of AO-based cancer therapy (Figure 1).
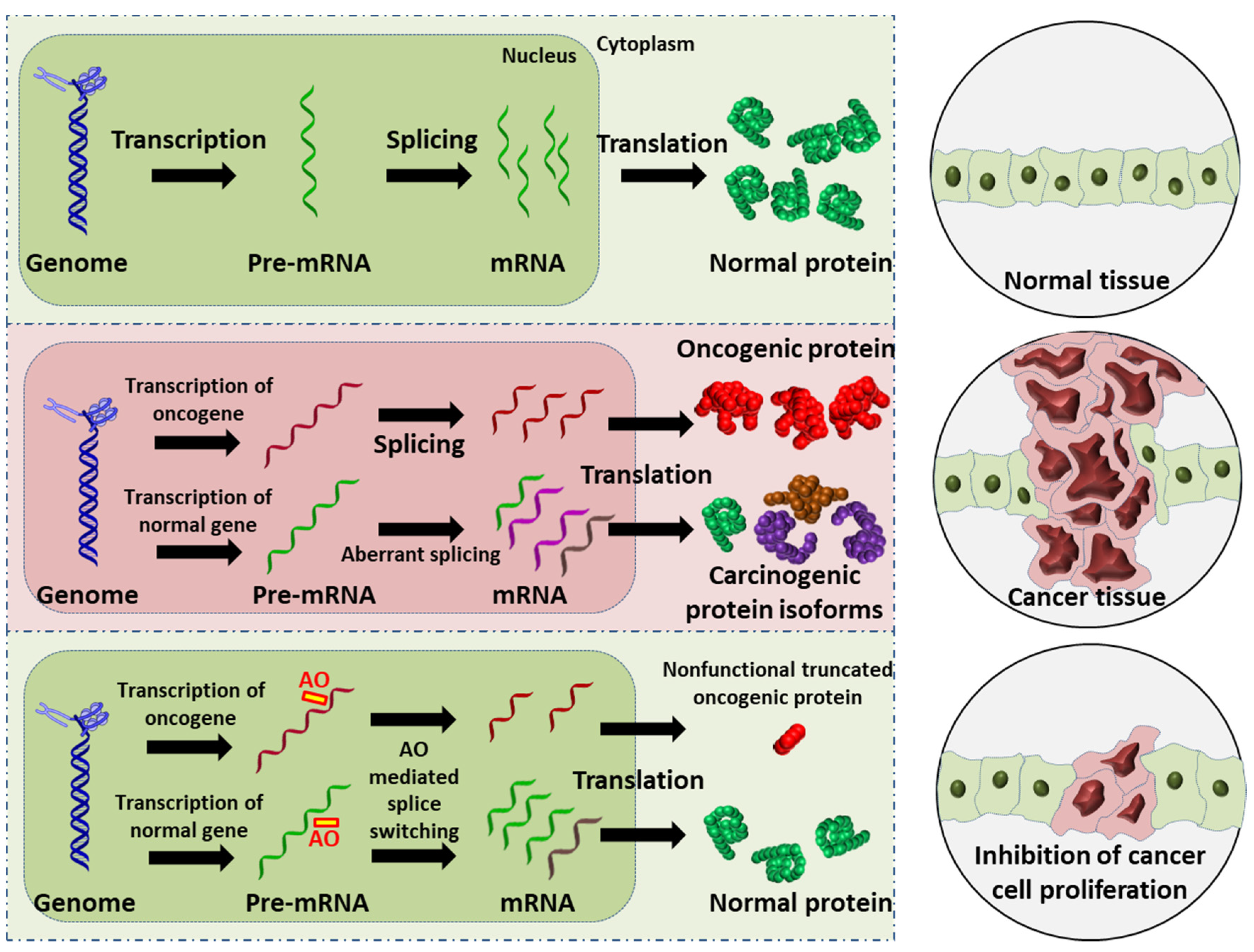 Figure 1. Schematic illustration of the AO-induced anticancer effect. (Upper): normal cell and tissue. (Middle): activation of oncogene leads to production of oncogenic proteins, and the aberrant splicing of normal gene results in splice variants that are translated into carcinogenic protein isoforms; these carcinogenic proteins collectively promote cancer cell proliferation. (Lower): AO-mediated splice switching can not only correct the aberrant splicing, thus reducing the production of carcinogenic protein isoforms (this is the focus of the present review), but can also induce premature stop codons in oncogenic mRNA that results in the synthesis of largely truncated, nonfunctional oncogenic proteins (this is a potential anticancer strategy). Collectively, AO intervention can inhibit cancer cell proliferation.
Figure 1. Schematic illustration of the AO-induced anticancer effect. (Upper): normal cell and tissue. (Middle): activation of oncogene leads to production of oncogenic proteins, and the aberrant splicing of normal gene results in splice variants that are translated into carcinogenic protein isoforms; these carcinogenic proteins collectively promote cancer cell proliferation. (Lower): AO-mediated splice switching can not only correct the aberrant splicing, thus reducing the production of carcinogenic protein isoforms (this is the focus of the present review), but can also induce premature stop codons in oncogenic mRNA that results in the synthesis of largely truncated, nonfunctional oncogenic proteins (this is a potential anticancer strategy). Collectively, AO intervention can inhibit cancer cell proliferation.
2. Exon-Skipping AOs in Cancer
2.1. Breast Cancer
2.2. Leukemia
| No. | Research | Ref. | |
|---|---|---|---|
| 1 | AO | SSO111 is a 20mer fully modified 2′-MOE-PS AO-targeting oncogene HER2. SSO111 induced exon 15 skipping during splicing, leading to the generation of a novel mRNA transcript that excludes exon 15.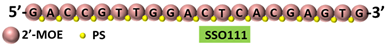 |
[25] |
| Mechanism | 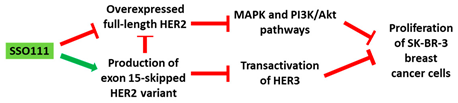 |
||
| 2 | AO | Acr-PNA 2794 is a 15mer fully modified PNA AO conjugated with Acr targeting HER2. Acr-PNA 2794 induced exon-19 skipping, leading to the generation of a novel mRNA transcript that excludes exon-19.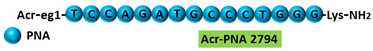 |
[26] |
| Mechanism | 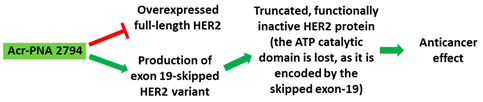 |
||
| 3 | AO | SSOe26 is a 15mer LNA-modified mixmer AO targeting HER4. SSOe26 induced exon 26 skipping, leading to the generation of a novel mRNA transcript that excludes exon 26 (CYT2 isoform).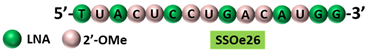 |
[29] |
| Mechanism | 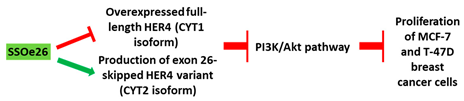 |
||
| 4 | AO | ASWT1exon5 is a 20mer 2′-MOE-PS gapmer AO targeting oncogene WT1. It induces RNase H-mediated degradation of exon 5-containing transcripts, thus increasing the proportion of transcripts that exclude exon 5. |
[35] |
| Mechanism | 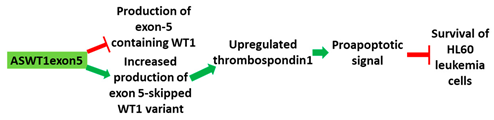 |
||
| 5 | AO | PNA 4577, 4578, 4580, and 4581 are 16mer fully modified PNA AOs conjugated with octaarginine or cholic acid-targeting oncogene TdT. These four PNAs all induced intron 7 retention, leading to the generation of a novel mRNA transcript that included intron-7.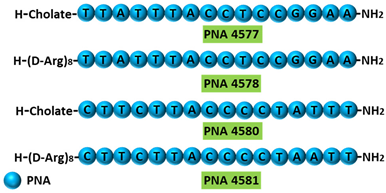 |
[39] |
| Mechanism | 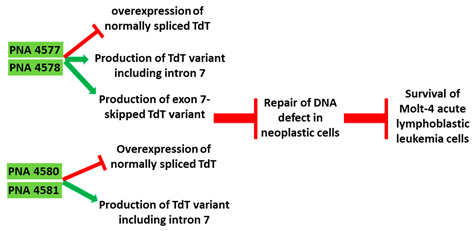 |
||
| 6 | AO | Morpholino MDM4 is a 25mer fully modified PMO AO targeting MDM4. Morpholino MDM4 induced exon 6 skipping, leading to nonsense-mediated decay of the mRNA transcript that excludes exon-6. |
[40] |
| Mechanism |  |
||
2.3. Melanoma
3. Conclusions
References
- Fackenthal, J.D.; Godley, L.A. Aberrant RNA splicing and its functional consequences in cancer cells. Dis. Models Mech. 2008, 1, 37–42.
- Johnson, M.B.; Kawasawa, Y.I.; Mason, C.E.; Krsnik, Ž.; Coppola, G.; Bogdanović, D.; Geschwind, D.H.; Mane, S.M.; State, M.W.; Šestan, N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 2009, 62, 494–509.
- Siva, K.; Covello, G.; Denti, M.A. Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic Acid Ther. 2014, 24, 69–86.
- Johnson, J.M.; Castle, J.; Garrett-Engele, P.; Kan, Z.; Loerch, P.M.; Armour, C.D.; Santos, R.; Schadt, E.E.; Stoughton, R.; Shoemaker, D.D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302, 2141–2144.
- Clark, T.A.; Schweitzer, A.C.; Chen, T.X.; Staples, M.K.; Lu, G.; Wang, H.; Williams, A.; Blume, J.E. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007, 8, R64.
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268.
- Dhir, A.; Buratti, E. Alternative splicing: Role of pseudoexons in human disease and potential therapeutic strategies. FEBS J. 2010, 277, 841–855.
- Weir, B.; Zhao, X.; Meyerson, M. Somatic alterations in the human cancer genome. Cancer Cell 2004, 6, 433–438.
- Mendelsohn, J.; Howley, P.; Israel, M.; Gray, J.; Thompson, C. The Molecular Basis of Cancer; Saunders Elsevier: Philadelphia, PA, USA, 2008.
- Wang, B.-D.; Lee, N.H. Aberrant RNA splicing in cancer and drug resistance. Cancers 2018, 10, 458.
- Gleave, M.E.; Monia, B.P. Antisense therapy for cancer. Nat. Rev. Cancer 2005, 5, 468–479.
- Arechavala-Gomeza, V.; Khoo, B.; Aartsma-Rus, A. Splicing Modulation Therapy in the Treatment of Genetic Diseases. Appl. Clin. Genet. 2014, 7, 245–252.
- Chen, S.; Sbuh, N.; Veedu, R.N. Antisense oligonucleotides as potential therapeutics for Type 2 Diabetes. Nucleic Acid Ther. 2021, 31, 39–57.
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416.
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321.
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694.
- Le, B.T.; Raguraman, P.; Kosbar, T.R.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Antisense oligonucleotides targeting angiogenic factors as potential cancer therapeutics. Mol. Ther.-Nucleic Acids 2019, 14, 142–157.
- Kushner, D.M.; Silverman, R.H. Antisense cancer therapy: The state of the science. Curr. Oncol. Rep. 2000, 2, 23–30.
- Dean, N.M.; Bennett, C.F. Antisense oligonucleotide-based therapeutics for cancer. Oncogene 2003, 22, 9087–9096.
- Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 2002, 277, 49374–49382.
- Ziegler, R.G.; Hoover, R.N.; Pike, M.C.; Hildesheim, A.; Nomura, A.M.; West, D.W.; Wu-Williams, A.H.; Kolonel, L.N.; Horn-Ross, P.L.; Rosenthal, J.F. Migration patterns and breast cancer risk in Asian-American women. JNCI J. Natl. Cancer Inst. 1993, 85, 1819–1827.
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182.
- Swain, S.M.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734.
- Moasser, M.M. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene 2007, 26, 6577–6592.
- Wan, J.; Sazani, P.; Kole, R. Modification of HER2 pre-mRNA alternative splicing and its effects on breast cancer cells. Int. J. Cancer 2009, 124, 772–777.
- Pankratova, S.; Nielsen, B.N.; Shiraishi, T.; Nielsen, P.E. PNA-mediated modulation and redirection of Her-2 pre-mRNA splicing: Specific skipping of erbB-2 exon 19 coding for the ATP catalytic domain. Int. J. Oncol. 2010, 36, 29–38.
- Menard, S.; Casalini, P.; Campiglio, M.; Pupa, S.; Agresti, R.; Tagliabue, E. HER2 overexpression in various tumor types, focussing on its relationship to the development of invasive breast cancer. Ann. Oncol. 2001, 12, S15–S19.
- Dias, N.; Stein, C. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355.
- Nielsen, T.O.; Sorensen, S.; Dagnaes-Hansen, F.; Kjems, J.; Sorensen, B. Directing HER4 mRNA expression towards the CYT2 isoform by antisense oligonucleotide decreases growth of breast cancer cells in vitro and in vivo. Br. J. Cancer 2013, 108, 2291–2298.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424.
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441.
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577.
- Lyu, X.; Xin, Y.; Mi, R.; Ding, J.; Wang, X.; Hu, J.; Fan, R.; Wei, X.; Song, Y.; Zhao, R.Y. Overexpression of Wilms tumor 1 gene as a negative prognostic indicator in acute myeloid leukemia. PLoS ONE 2014, 9, e92470.
- Menssen, H.; Renkl, H.; Rodeck, U.; Maurer, J.; Notter, M.; Schwartz, S.; Reinhardt, R.; Thiel, E. Presence of Wilms’ tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia 1995, 9, 1060–1067.
- Renshaw, J.; Orr, R.M.; Walton, M.I.; Te Poele, R.; Williams, R.D.; Wancewicz, E.V.; Monia, B.P.; Workman, P.; Pritchard-Jones, K. Disruption of WT1 gene expression and exon 5 splicing following cytotoxic drug treatment: Antisense down-regulation of exon 5 alters target gene expression and inhibits cell survival. Mol. Cancer Ther. 2004, 3, 1467–1484.
- Miwa, H.; Beran, M.; Saunders, G. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 1992, 6, 405–409.
- Bollum, F.J. Terminal deoxynucleotidyl transferase as a hematopoietic cell marker. Blood 1979, 54, 1203–1215.
- Farahat, N.; Lens, D.; Morilla, R.; Matutes, E.; Catovsky, D. Differential TdT expression in acute leukemia by flow cytometry: A quantitative study. Leukemia 1995, 9, 583–587.
- Montazersaheb, S.; Kazemi, M.; Nabat, E.; Nielsen, P.E.; Hejazi, M.S. Downregulation of TdT expression through splicing modulation by antisense peptide nucleic acid (PNA). Curr. Pharm. Biotechnol. 2019, 20, 168–178.
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A. Antisense oligonucleotide–mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84.
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications, Brisbane, Australia, 2017.
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247.

