 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zheng Dong | + 1785 word(s) | 1785 | 2021-11-17 08:48:56 | | | |
| 2 | Catherine Yang | Meta information modification | 1785 | 2021-11-24 06:41:10 | | |
Video Upload Options
Cisplatin is a potent chemotherapy drug used for the treatment of various types of tumors, but it has remarkable side effects or toxicity in normal tissues. The kidney is highly vulnerable to cisplatin toxicity due to the accumulation of cisplatin in renal tubule cells. Acute kidney injury occurs in 20–30% of patients and manifests as kidney cell death, tissue damage, rapid loss of renal function or renal failure, and even death.
1. Introduction
Cisplatin is a potent chemotherapy drug used for the treatment of various types of tumors [1], but it has remarkable side effects or toxicity in normal tissues [2]. The kidney is highly vulnerable to cisplatin toxicity due to the accumulation of cisplatin in renal tubule cells [3]. Acute kidney injury occurs in 20–30% of patients and manifests as kidney cell death, tissue damage, rapid loss of renal function or renal failure, and even death [4]. Following cisplatin chemotherapy, a significant portion of cancer patients develop chronic kidney problems [5]. Therefore, it is vital to explore the mechanism of the nephrotoxicity of cisplatin and identify preventive or protective measures.
Autophagy is a lysosomal degradation pathway that clears dysfunctional or obsolete cytoplasmic components [6][7]. In the kidney, the basal level of autophagy plays a role in maintaining renal cell homeostasis and function under normal physiological conditions. Autophagy is induced in response to cellular stress when the kidney is diseased or exposed to insults or toxins, such as cisplatin. Autophagy is generally considered pivotal in promoting cell survival and protecting against acute cisplatin nephrotoxicity [8][9][10][11]. Autophagy also participates in the regulation of maladaptive kidney repair and renal fibrosis after acute kidney injury and during the progression of chronic kidney disease [12][13][14][15][16][17]. However, little is known about the role and regulation of autophagy in the development of chronic kidney problems after cisplatin exposure.
In cancers, the role of autophagy is Janus-faced. On the one hand, it may limit or prevent tumorigenesis, but, on the other hand, autophagy may reduce the efficacy of cancer therapy by promoting cancer cell survival [18]. Autophagy induction by cisplatin is associated with the development of cisplatin resistance in many types of cancer cells, including bladder cancer, esophageal cancer [19], lung cancer [20], ovarian cancer [21], and bone cancer [22]. Moreover, the association between the loss of autophagy genes and the decrease in other tumors’ sensitivity to chemotherapy drugs further highlights the significance of autophagy in cancer suppression in some tumors [23]. Therefore, autophagy upregulation can protect the kidneys against acute cisplatin injury, but its effect on cancers is context-dependent. When targeting autophagy as a strategy for kidney protection, the effect on tumors must be considered.
2. Cisplatin Nephrotoxicity
Cisplatin is efficacious for solid tumor treatment, either alone or in combination with other therapies, but it also causes toxicity in multiple organs and tissues, especially in the kidney [1][24]. Approximately 20–30% of patients who receive cisplatin develop acute kidney injury (AKI), which is characterized by rapid loss of renal function, the accumulation of end products of nitrogen metabolism, and the disturbance of water and electrolytes [25]. The long-term effects of cisplatin on the kidney are not entirely understood. Brillet et al. [26] and Latcha et al. [5] reported that cisplatin might lead to a subclinical but persistent low glomerular filtration rate. Therefore, a thorough understanding of AKI and CKD induced by cisplatin is necessary for clinical treatment.
Cisplatin nephrotoxicity is a multifactorial process including various pathophysiological events, such as microvascular disorders, tubular injury, tubular cell death, and inflammatory response. Among them, tubular cell injury and death is a critical pathological feature [27][28][29]. In experimental models of AKI, the proximal tubule, especially the S3 segment, is highly sensitive and most vulnerable to damage [30][31]. The renal tubule damages caused by cisplatin are often manifested as tubule dilation, cast formation, and tubular cell apoptosis and necrosis [32][33].
Many studies have focused on the cellular and molecular mechanisms of tubular damage, especially proximal tubular injury. Renal tubular cells mainly uptake cisplatin through organic cation transporters 2 (OCT2) [34] and copper transporter 1 (CTR1) [35]. After entry into the cell, cisplatin may undergo a series of bio-activation processes to generate various toxic metabolites, which are catalyzed by γ-glutamyl transpeptidase (GGT) and cysteine-S-conjugate β-lyase [36]. Then, it may bind to DNA, leading to inter- and intrastrand cross-links [37], causing DNA damage and DNA-damage response [38][39], resulting in cell cycle arrest and cell death [40]. Multiple signaling pathways are activated upon cisplatin exposure, such as mitogen-activated protein kinases (MAPKs) [41] and the p53-DNA damage response pathway [42][43], leading to apoptosis, necroptosis [44], and ferroptosis [45]. Cisplatin also evokes oxidative stress [46] , ER stress [47], mitochondrial dysfunction [48].
Currently, a common regimen of cisplatin treatment is weekly administration of relatively low doses of cisplatin for several cycles. While this regimen reduces the side effects of cisplatin, a significant portion of patients still develop acute kidney injury, and some progress into chronic kidney disease. Repeated low-dose cisplatin (RLDC) treatment models were established to study chronic kidney problems after cisplatin treatment.
3. Autophagy in Cisplatin-Induced AKI
ER stress is induced along with autophagy after both cisplatin exposure [49][50] and ischemia-reperfusion (IR) injury [51]. Whether autophagy induction is related to ER stress was confirmed by pharmacological approaches. For example, inhibitors of ER stress suppressed cyclosporine-induced autophagy [50] and activators of ER stress notably increased autophagy in renal proximal tubular cells [52]. Our recent study has further demonstrated a reciprocal regulation between ER stress and autophagy in renal tubular cells. Specifically, ER stress induces autophagy leading to fibrotic changes in renal tubular cells, whereas autophagy, upon activation, reduces ER stress in these cells providing a negative feedback mechanism. ER stress might affect the induction of autophagy via regulating calcium release, REDD1/mTOR, and AKT/mTOR pathways. Moreover, the autophagy machinery components are tightly regulated by unfolded protein response (UPR) [53]. Gozuacik et al. [54] showed that death-associated protein kinase1 (DAPK1) was induced by ER stress and regulated autophagy via the phosphorylation of Beclin 1, and autophagy was inhibited in Dapk- null cells in tunicamycin-induced kidney injury. Conceivably, moderate ER stress activates cytoprotective mechanisms such as UPR and autophagy, whereas severe ER stress leads to irreversible cell damage and cell death. Delineating the molecular basis of this shift may lead to new strategies for reducing nephrotoxicity in cisplatin chemotherapy.
It is well recognized that nutrient or energy deprivation is a core reason for the induction of autophagy, which is mainly mediated by mTOR, AMPK, and nicotinamide adenine dinucleotide (NAD+) metabolism.
mTORC1 suppresses autophagy by phosphorylating ULK1 and ATG13 to inhibit the ULK1–ULK2 complex formation and autophagy initiation. Several notable autophagy regulators may work by modulating mTOR complex 1 (mTORC1). AMPK may phosphorylate ULK1 to induce autophagy directly or phosphorylate mTORC1 to induce autophagy indirectly. Growth factors regulate mTORC1 activity mainly by activating two classical pathways of mTORC1, PI3K/AKT/mTORC1, and Ras/Raf/MEK/ERK/mTORC1 signaling pathways [55]. In cisplatin AKI, several factors may regulate autophagy through mTORC1. For example, p53 may activate autophagy through the inhibition of mTORC1 by AMPK [56] or miR-199a-3p [57], while activation of protein kinase Cδ (PKCδ) in cisplatin AKI inhibits autophagy through the AKT/mTORC1/ULK1 pathway [58]. Cisplatin-induced NQO1 may inhibit autophagy through activation of the AMPK/TSC2/ mTORC1 signaling pathway [59]. In addition, histone deacetylase (HDAC) inhibitors were shown to protect against cisplatin-induced kidney tubular cell injury [60]. More recent work showed that HDAC inhibitors may protect kidney tubular cells by enhancing autophagy at least partially by suppressing mTORC1 [61]. Well-known inhibitors of mTORC1, rapamycin, and everolimus both affect the cisplatin kidney injury and repair. Together, these studies indicate a role of mTORC1 and related signaling in the regulation of autophagy in cisplatin nephrotoxicity.
In addition to the energy signaling pathway, other pathways also contribute to the regulation of autophagy. For example, active mitogen-activated protein kinase 8 (MAPK8; also known as JNK1) [62] and death-associated protein kinases (DAP kinases) [63] can activate Beclin 1 to promote autophagy. Some epigenetic mechanisms, kinases, and transcriptional factors are also involved in the regulation of the elongation and fusion stages of autophagy. For example, eukaryotic translation initiation factor 2 subunit-α (eIF2α) [64] and NF-κB kinase inhibitor (IKK) [65] can induce autophagy by regulating some ATG genes. Transcription factor EB (encoded by TFEB) [66] and nuclear export of zinc-finger protein with KRAB and SCAN domains 3 (encoded byZKSCAN3) [67] regulate autophagy by transcriptional regulation of autophagy/lysosomes genes. In cisplatin-induced AKI, activation of TFEB-mediated autophagy and attenuation of mitochondrial dysfunction by trehalose might depend on the inhibition of Akt [68].
4. Strategies Targeting Autophagy in Cisplatin Nephrotoxicity during Chemotherapy
Given the critical role of autophagy in cisplatin-induced kidney disease, the pharmacological intervention of autophagy should be a meaningful strategy for the prevention and treatment of kidney diseases during cisplatin chemotherapy. In tumors, autophagy has long been considered a double-edged sword in tumorigenesis and cancer therapy. In the kidney, autophagy is protective during acute cisplatin nephrotoxicity, but the role of autophagy in chronic kidney problems following cisplatin chemotherapy is unclear. Any strategies targeting autophagy in cisplatin chemotherapy must be taken into consideration of the effects in both tumors and normal tissues, especially the kidneys ( Figure 1 ).
Besides the above, autophagy activation in some tumors helps the recognition of cancer cells by the immune system to facilitate the anti-tumor immune surveillance and promote autophagic cell death through various pathways of cell death, including apoptosis, necrosis, and necroptosis [69][70]. These effects imply that autophagy might be helpful for cancer prevention and treatment through reducing chromosome instability, increasing immune surveillance, and promoting cell death ( Figure 1 ).
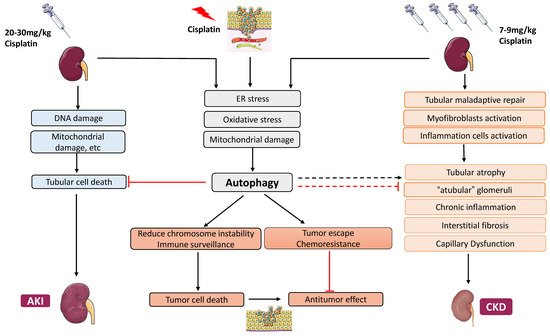
Before tumorigenesis, autophagy defects result in poor elimination of oncogenic proteins, toxic aggregates, and damaged organelles, leading to chronic inflammation, tissue damage, genome instability, and oncogenic gene activation [71][72]. However, autophagy also supports metabolism and survival in cancer cells once the tumor has formed [73]. In this case, autophagy protects cancer cells and increases their resistance to anticancer drugs, and inhibition of autophagy would sensitize cancer cells to therapy and enhance the therapeutic effect of chemotherapeutics.
From the current findings, many drugs that inhibit autophagy can decrease the resistance of tumors to cisplatin but can cause damage to acute kidney injury. The function of autophagy in chronic kidney problems following cisplatin treatment remains unclear, but there is evidence for the involvement of autophagy in the development of CKD after kidney injury. In this regard, although inhibiting autophagy aggravates acute kidney injury, it may protect the progression of chronic kidney injury. Therefore, it carefully avoids using autophagy inhibitors in patients with tumors and acute kidney injury, but it may be a promising option for patients with chronic kidney disease. Therefore, it is imperative to verify the role of autophagy in chronic kidney disease.
References
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378.
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA damage to cancer chemotherapy. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 67, 93–130.
- de Jonge, M.J.; Verweij, J. Renal Toxicities of Chemotherapy. Semin. Oncol. 2006, 33, 68–73.
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A. Acute Kidney Injury Network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31.
- Latcha, S.; Jaimes, E.A.; Patil, S.; Glezerman, I.G.; Mehta, S.; Flombaum, C.D. Long–Term Renal Outcomes after Cisplatin Treatment. Clin. J. Am. Soc. Nephrol. 2016, 11, 1173–1179.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741.
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075.
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508.
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791.
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031.
- Periyasamy-Thandavan, S.; Jiang, M.; Schoenlein, P.; Dong, Z. Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Physiol. 2009, 297, F244–F256.
- Nam, S.A.; Kim, W.-Y.; Kim, J.W.; Park, S.H.; Kim, H.L.; Lee, M.-S.; Komatsu, M.; Ha, H.; Lim, J.H.; Park, C.W.; et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019, 10, 1–10.
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J. Clin. Investig. 2019, 129, 2374–2389.
- Livingston, M.J.; Ding, H.-F.; Huang, S.; Hill, J.A.; Yin, X.-M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998.
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486.
- Baisantry, A.; Bhayana, S.; Rong, S.; Ermeling, E.; Wrede, C.; Hegermann, J.; Pennekamp, P.; Sörensen-Zender, I.; Haller, H.; Melk, A.; et al. Autophagy Induces Prosenescent Changes in Proximal Tubular S3 Segments. J. Am. Soc. Nephrol. 2015, 27, 1609–1616.
- Ding, Y.; Kim, S.L.; Lee, S.-Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy Regulates TGF-β Expression and Suppresses Kidney Fibrosis Induced by Unilateral Ureteral Obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846.
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838.
- O’Donovan, T.R.; O’Sullivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524.
- Sirichanchuen, B.; Pengsuparp, T.; Chanvorachote, P. Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol. Cell Biochem. 2012, 364, 11–18.
- Wang, J.; Wu, G.S. Role of Autophagy in Cisplatin Resistance in Ovarian Cancer Cells. J. Biol. Chem. 2014, 289, 17163–17173.
- Jiang, K.; Zhang, C.; Yu, B.; Chen, B.; Liu, Z.; Hou, C.; Wang, F.; Shen, H.; Chen, Z. Autophagic degradation of FOXO3a represses the expression of PUMA to block cell apoptosis in cisplatin-resistant osteosarcoma cells. Am. J. Cancer Res. 2017, 7, 1407–1422.
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42.
- Beyer, J.; Rick, O.; Weinknecht, S.; Kingreen, D.; Lenz, K.; Siegert, W. Nephrotoxicity after high-dose carboplatin, etoposide and ifosfamide in germ-cell tumors: Incidence and implications for hematologic recovery and clinical outcome. Bone Marrow Transplant. 1997, 20, 813–819.
- Campdera, F.J.G.; Gonzalez, P.; Carrillo, A.; Estelles, M.C.; Rengel, M. Cisplatin nephrotoxicity: Symptomatic hypomagnesemia and renal failure. Int. J. Pediatr. Nephrol. 1986, 7, 151–152.
- Brilleta, G.; Demya, G.; Jacquiauda, C.; Mignotb, L.; Bunkera, D.; Meilletc, D.; Raymond, F.; Jacobs, C.; Brillet, G.; Deray, G.; et al. Long-Term Renal Effect of Cisplatin in Man. Am. J. Nephrol. 1994, 14, 81–84.
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766.
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated Cell Death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701.
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307.
- Lieberthal, W.; Nigam, S.K. Acute Renal Failure. I. Relative importance of proximal vs. distal tubular injury. Am. J. Physiol. Physiol. 1998, 275, F623–F632.
- Lieberthal, W.; Nigam, S.K. Acute Renal Failure. II. Experimental models of acute renal failure: Imperfect but indispensable. Am. J. Physiol. Physiol. 2000, 278, F1–F12.
- Ozkok, A.; Edelstein, C.L. Pathophysiology of Cisplatin-Induced Acute Kidney Injury. BioMed Res. Int. 2014, 2014, 1–17.
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007.
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstädt, H.; Koepsell, H.; Piechota, H.-J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin Nephrotoxicity Is Critically Mediated via the Human Organic Cation Transporter 2. Am. J. Pathol. 2005, 167, 1477–1484.
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Physiol. 2009, 296, F505–F511.
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of Cisplatin to a Nephrotoxin in Proximal Tubule Cells. J. Am. Soc. Nephrol. 2003, 14, 1–10.
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320.
- Yan, M.; Tang, C.; Ma, Z.; Huang, S.; Dong, Z. DNA damage response in nephrotoxic and ischemic kidney injury. Toxicol. Appl. Pharmacol. 2016, 313, 104–108.
- Zhu, S.; Pabla, N.; Tang, C.; He, L.; Dong, Z. DNA damage response in cisplatin-induced nephrotoxicity. Arch. Toxicol. 2015, 89, 2197–2205.
- Ju, S.-M.; Kim, M.-S.; Jo, Y.-S.; Jeon, Y.-M.; Bae, J.-S.; Pae, H.-O.; Jeon, B.-H. Licorice and its active compound glycyrrhizic acid ameliorates cisplatin-induced nephrotoxicity through inactivation of p53 by scavenging ROS and overexpression of p21 in human renal proximal tubular epithelial cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 890–899.
- Francescato, H.D.; Costa, R.S.; Silva, C.G.; Coimbra, T.M. Treatment with a p38 MAPK inhibitor attenuates cisplatin nephrotoxicity starting after the beginning of renal damage. Life Sci. 2009, 84, 590–597.
- Jiang, M.; Dong, Z. Regulation and Pathological Role of p53 in Cisplatin Nephrotoxicity. J. Pharmacol. Exp. Ther. 2008, 327, 300–307.
- Wei, Q.; Dong, G.; Yang, T.; Megyesi, J.; Price, P.M.; Dong, Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Physiol. 2007, 293, F1282–F1291.
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658.
- Deng, F.; Sharma, I.; Dai, Y.; Yang, M.; Kanwar, Y.S. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J. Clin. Investig. 2019, 129, 5033–5049.
- Takai, N.; Abe, K.; Tonomura, M.; Imamoto, N.; Fukumoto, K.; Ito, M.; Momosaki, S.; Fujisawa, K.; Morimoto, K.; Takasu, N.; et al. Imaging of reactive oxygen species using hydromethidine in mice with cisplatin-induced nephrotoxicity. EJNMMI Res. 2015, 5, 38.
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390.
- Zsengellér, Z.K.; Ellezian, L.; Brown, D.; Horváth, B.; Mukhopadhyay, P.; Kalyanaraman, B.; Parikh, S.M.; Karumanchi, S.A.; Stillman, I.E.; Pacher, P. Cisplatin Nephrotoxicity Involves Mitochondrial Injury with Impaired Tubular Mitochondrial Enzyme Activity. J. Histochem. Cytochem. 2012, 60, 521–529.
- Rovetta, F.; Stacchiotti, A.; Consiglio, A.; Cadei, M.; Grigolato, P.G.; Lavazza, A.; Rezzani, R.; Aleo, M.F. ER signaling regulation drives the switch between autophagy and apoptosis in NRK-52E cells exposed to cisplatin. Exp. Cell Res. 2012, 318, 238–250.
- Pallet, N.; Bouvier, N.; Legendre, C.; Gilleron, J.; Codogno, P.; Beaune, P.; Thervet, E.; Anglicheau, D. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 2008, 4, 783–791.
- Chandrika, B.B.; Yang, C.; Ou, Y.; Feng, X.; Muhoza, D.; Holmes, A.F.; Theus, S.; Deshmukh, S.; Haun, R.S.; Kaushal, G.P. Endoplasmic Reticulum Stress-Induced Autophagy Provides Cytoprotection from Chemical Hypoxia and Oxidant Injury and Ameliorates Renal Ischemia-Reperfusion Injury. PLoS ONE 2015, 10, e0140025.
- Kawakami, T.; Inagi, R.; Takano, H.; Sato, S.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol. Dial. Transplant. 2009, 24, 2665–2672.
- Deegan, S.; Saveljeva, S.; Gorman, A.; Samali, A. Stress-induced self-cannibalism: On the regulation of autophagy by endoplasmic reticulum stress. Cell. Mol. Life Sci. 2012, 70, 2425–2441.
- Gozuacik, D.; Bialik, S.; Raveh, T.; Mitou, G.; Shohat, G.; Sabanay, H.; Mizushima, N.; Yoshimori, T.; Kimchi, A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008, 15, 1875–1886.
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371.
- Wei, L.; Chen, W.; Zou, Y.; Huang, H.; Pan, B.; Jin, S.; Huang, R.; Nie, S.; Kong, G. AMP-activated protein kinase regulates autophagic protection against cisplatin-induced tissue injury in the kidney. Genet. Mol. Res. 2015, 14, 12006–12015.
- Yang, A.; Liu, F.; Guan, B.; Luo, Z.; Lin, J.; Fang, W.; Liu, L.; Zuo, W. p53 induces miR-199a-3p to suppress mechanistic target of rapamycin activation in cisplatin-induced acute kidney injury. J. Cell. Biochem. 2019, 120, 17625–17634.
- Zhang, D.; Pan, J.; Xiang, X.; Liu, Y.; Dong, G.; Livingston, M.J.; Chen, J.-K.; Yin, X.-M.; Dong, Z. Protein Kinase Cδ Suppresses Autophagy to Induce Kidney Cell Apoptosis in Cisplatin Nephrotoxicity. J. Am. Soc. Nephrol. 2016, 28, 1131–1144.
- Kim, T.-W.; Kim, Y.-J.; Kim, H.-T.; Park, S.-R.; Lee, M.-Y.; Park, Y.-D.; Lee, C.-H.; Jung, J.-Y. NQO1 Deficiency Leads Enhanced Autophagy in Cisplatin-Induced Acute Kidney Injury Through the AMPK/TSC2/mTOR Signaling Pathway. Antioxid. Redox Signal. 2016, 24, 867–883.
- Dong, G.; Luo, J.; Kumar, V.; Dong, Z. Inhibitors of histone deacetylases suppress cisplatin-induced p53 activation and apoptosis in renal tubular cells. Am. J. Physiol. Physiol. 2010, 298, F293–F300.
- Liu, J.; Livingston, M.J.; Dong, G.; Tang, C.; Su, Y.; Wu, G.; Yin, X.-M.; Dong, Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018, 9, 1–15.
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688.
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292.
- B’Chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699.
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2009, 29, 619–631.
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia-Arencibia, M.; Vetrini, F.; Erdin, S.; Huynh, T.; Medina, D.L.; Colella, P.; Sardiello, M.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433.
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. ZKSCAN3 Is a Master Transcriptional Repressor of Autophagy. Mol. Cell 2013, 50, 16–28.
- Zhu, L.; Yuan, Y.; Yuan, L.; Li, L.; Liu, F.; Liu, J.; Chen, Y.; Lu, Y.; Cheng, J. Activation of TFEB-mediated autophagy by trehalose attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Theranostics 2020, 10, 5829–5844.
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577.
- Potočnjak, I.; Šimić, L.; Vukelić, I.; Domitrović, R. Oleanolic acid attenuates cisplatin-induced nephrotoxicity in mice and chemosensitizes human cervical cancer cells to cisplatin cytotoxicity. Food Chem. Toxicol. 2019, 132, 110676.
- Chen, H.-Y.; White, E. Role of Autophagy in Cancer Prevention. Cancer Prev. Res. 2011, 4, 973–983.
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64.
- Guo, J.Y.; Xia, B.; White, E. Autophagy-Mediated Tumor Promotion. Cell 2013, 155, 1216–1219.


