Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rodrigo Araldi | + 3751 word(s) | 3751 | 2021-10-13 08:49:50 | | | |
| 2 | Lindsay Dong | Meta information modification | 3751 | 2021-11-19 16:42:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Araldi, R. Cancer-Derived Exosomes in Carcinogenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/16164 (accessed on 28 July 2026).
Araldi R. Cancer-Derived Exosomes in Carcinogenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/16164. Accessed July 28, 2026.
Araldi, Rodrigo. "Cancer-Derived Exosomes in Carcinogenesis" Encyclopedia, https://encyclopedia.pub/entry/16164 (accessed July 28, 2026).
Araldi, R. (2021, November 18). Cancer-Derived Exosomes in Carcinogenesis. In Encyclopedia. https://encyclopedia.pub/entry/16164
Araldi, Rodrigo. "Cancer-Derived Exosomes in Carcinogenesis." Encyclopedia. Web. 18 November, 2021.
Copy Citation
The exosome-mediated crosstalk between cancer and non-cancer cells within the tumor microenvironment (TME) contributes to the acquisition of all hallmarks of cancer and leads to the formation of cancer stem cells (CSCs), which exhibit resistance to a range of anticancer drugs.
exosomes
cancer
tumor microenvironment (TME)
immunomodulation
epithelial-mesenchymal transition (EMT)
mesenchymal-stem cell (MSC)
cell-free therapy
1. Exosome Biogenesis
Naturally, all cell types produce and secrete different types of extracellular vesicles (EVs), which participate in both physiological and pathophysiological processes [1][2]. Depending on their size, biogenesis mechanisms, or function, these vesicles are classified as microvesicles (100–1000 nm), exosomes (30–200 nm), or apoptotic bodies (generally > 1000 nm) [3][4][5].
Typically, exosomes are surrounded by a phospholipid membrane containing an abundance of cholesterol, sphingomyelin, ceramide, lipid rafts, and evolutionarily conserved biomarkers, which are used to distinguish them from microvesicles or apoptotic bodies, such as tetraspanins (CD9, CD63, CD81, and CD82), heat shock proteins (Hsp60, 70, and 90), major histocompatibility component classes I (MHC-I) and II (MHC-II), Alix, Tsg101, lactadherin, and lysosome-associated membrane glycoprotein 2, as illustrated in Figure 1 [3][6][7][8][9][10]. Besides these proteins, exosomes contain specific proteins and transcripts, which are responsible for eliciting the regulation of recipient cells.
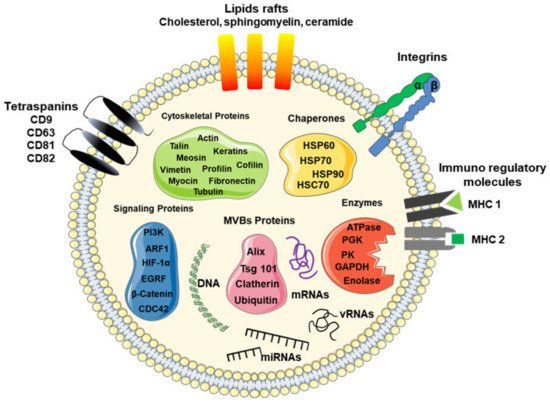
Figure 1. Schematic model of a typical exosome. The model shows a nanosized membrane-bound extracellular vesicle, with a diameter between 30 and 200 nm, expressing several proteins as a marker for exosomes, including tetraspanins (CD9, CD63, and CD81), Alix, Tsg101, and heat shock proteins (HSP-60, -70, and -90), as well as surface proteins, such as tetraspanins, integrins, immunoregulatory proteins (MHC-I and MHC-II), cytoskeletal proteins, signaling proteins, enzymes, and nucleic acids, such as coding RNAs (mRNAs) and non-coding RNAs (miRNAs and lncRNAs).
2. Molecular Cargo
Exosomes contain selective repertoires of proteins, nucleic acids (RNAs), lipids, and metabolites that regulate signaling pathways in the recipient cells [11]. The enrichment of a particular set of molecules within the exosomes suggests the existence of specific sorting mechanisms that orchestrate the selective packaging of the RNAs and proteins [11].
For many years, the sorting mechanism remained unclear. However, nowadays, it is clear that the selective packaging of RNAs and proteins is governed by the endosomal sorting complex required for transport (ESCRT), which also contributes to exosome formation.
The ESCRT is protein machinery composed of four ESCRT proteins (ESCRT-0, -I, -II, and -III) that work cooperatively to facilitate MVB formation, vesicle budding, and protein cargo sorting [12][13].
The ESCRT-mediated sorting is initiated by recognition and sequestration of ubiquitinated proteins to specific domains (the Hrs FYVE domain with phosphatidylinositol 3-phosphate (PtdIns3P)) of the endosomal membrane via ubiquitin-binding subunits of ESCRT-0 [12][14]. Next, the Hrs PSAP domain of the ESCRT-0 interacts with the subunit tumor susceptibility gene 101 (tsg101) of ESCRT-I [12][14]. ESCRT-I recruits the ESCRT-II proteins, which recruit and activate the ESCRT-III complex, which promotes the budding processes [12][14]. This occurs because the Snf7 protein of the ESCRT-III complex forms oligomeric assemblies, promoting vesicle budding [12][14]. Snf7 also recruits the Alix protein, stabilizing the ESCRT-III assembly [12][14]. Following cleaving the buds to form ILVs, the ESCRT-III complex separates from the MVB membrane with energy supplied by the sorting protein ATP Vps4 [12].
Although ESCRT-III is considered to be required for the scission of the ILVs into the MVE lumen [15], studies have reported the presence of ILVs within the lumen of MVBs in the ESCRT-depleted cells, indicating that ESCRT-independent pathways for ILV formation exist [16][17].
In this sense, recent evidence supports an alternative pathway for sorting exosomal cargo into MVBs in an ESCRT-independent manner, which seems to depend on raft-based microdomains for the lateral segregation of cargo within the endosomal membrane [12][16]. These microdomains are highly enriched in sphingomyelinases, from which ceramides can be formed by hydrolytic removal of the phosphocholine moiety [12][18].
The cone-shaped structure of ceramides might cause spontaneous negative curvature of the endosomal membrane, thereby promoting domain-induced budding [12][18].
3. Cancer-Derived Exosomes in Carcinogenesis
Cells of different tissue types produce and release exosomes to facilitate intercellular communication [19]. For this reason, it is not surprising that cancer-derived exosomes mediate the communication between cancer cells and non-cancer cells within the TME as well as malignant and non-malignant cells, regulating all carcinogenesis steps [20].
Typically, exosomes derived from cancer cells are larger than those derived from non-cancer cells. This size difference can be attributed to the heterogeneous nature of cancer cells, since different subclones of cancer cells are present within the TME, as well as the overexpression of genes related to the carcinogenic process [21]. For this reason, exosomes derived from cancer cells have been referred to as oncosomes (100–400 nm) or large oncosomes (LOs, 1–10 μm) according to their size and cargoes, as illustrated in Figure 2 [21]. Oncosomes are vesicles carrying abnormal and transforming macromolecules, including oncoproteins [21][22]. LOs are atypical extracellular vesicles, produced as a byproduct of non-apoptotic plasma membrane blebbing from cancer cells, and induced by silencing of the cytoskeletal regulator Diaphanous-related formin-3 (DIAPH3), by overexpression of the oncoproteins MyrAKT1, HB-EGF, and caveolin-1, or by the activation of the EGFR [23][24].
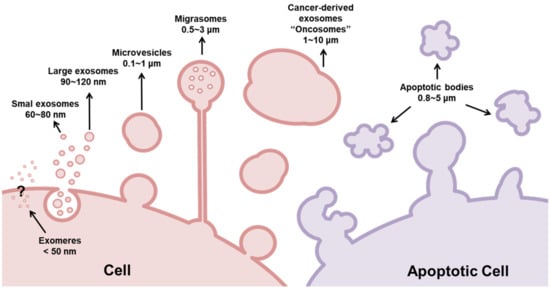
Figure 2. Classification of extracellular vesicles (EVs) according to their size. Basically, EVs are classified as exosomes (30–150 nm), microvesicles (100–1000 nm), and apoptotic bodies (800–5000 nm). While microvesicles and exosomes can operate as ‘safe containers’ mediating intercellular communication, apoptotic bodies appear after the disassembly of an apoptotic cell into subcellular fragments. Although they were previously regarded as garbage bags, emerging evidence supports the view that the apoptotic bodies are capable of delivering useful materials to healthy recipient cells. Different from exosomes, microvesicles are generated from the direct outward blebbing and pinching of the plasma membrane. Similar to exosomes, these vesicles carry proteins, nucleic acids, and bioactive lipids to recipient cells; however, they are larger than exosomes. Exosomes are conserved structures that originate as intraluminal vesicles during the assembly of multivesicular bodies, mediating cell-to-cell communication. However, current studies show that cancer-derived exosomes are larger than those secreted by normal/healthy cells. For this reason, these nanosized EVs were subclassified as exomers (<50 nm), small exosomes (60–80 nm), large exosomes (90–120 nm), and oncosomes (100–10,000 nm). Recently, a novel type of EV was described: migrasomes (500–3000 nm). Migrasomes are vesicular structures that mediate migracytocis, a cell migration mechanism mediated by these EVs.
3.1. Cancer-Derived Exosomes Mediate Crosstalk between Inflammation and Cancer Initiation
Cancer initiation is characterized by irreversible genetic alterations (driver mutation) that lead to the gain of function of oncogenes and/or loss of tumor suppression genes [25]. In addition, these mutations, associated with mitogenic stimuli from pre-cancerous micromilieu (cancer promotion), induce “initiated” cell proliferation (cancer progression). These combined steps increase the genomic instability, facilitating the novel mutations during the somatic evolution (passenger mutation) [26].
Current studies have demonstrated that exosomes are a key mediator of intercellular communication between cancer cells and non-cancer cells within the TME, acting as initiators of carcinogenesis by mediating crosstalk between inflammation and cancer initiation [27][28][29].
Both historically and contemporarily, cancer has been seen as an inflammatory disease [30][31]. However, in the last couple of decades, the contribution of the immune system and inflammation to cancer development has gained an enormous amount of interest [31]. This interest has allowed us to confirm that inflammation predisposes to the development of cancer and contributes to the acquisition of many hallmarks of cancer [31][32][33][34].
In this sense, studies have shown that exosomes produced and released by cancer cells contain various biomolecules, including nuclear factor kappa B (NFκB) and signal transducer activator of transcriptions 3 (STAT3), as well as inflammatory cytokines, such as interleukin (1L)-1β, -6, and tumor necrosis factor-alpha (TNF-α), which promotes the recruitment of immune cells to target sites as revisited by Othman et al. [35].
In 2013, Bretz et al. [36] showed that exosomes obtained from malignant ascites of ovarian cancer patients were able to bind to Toll-like receptors (TLR2 and TLR4) present on the surface of THP-1 cells (a spontaneously immortalized human monocyte-like cell line), inducing the production and secretion of the pro-inflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α in a NFκB- and STAT3-dependent manner.
However, the cancer-derived exosomes’ action is not limited to monocyte recruitment. Studies already demonstrated that breast [37] and gastric cancer-derived exosomes induce the differentiation of monocytes into M1 macrophages in a NFκB-dependent manner, stimulating the production of pro-inflammatory cytokines (GCSF, IL-6, IL-8, IL-1β, CCL2, and TNF-α) [38]. Interestingly, Chow et al. [37] revealed that the activation of NFκB in monocytes/macrophages occurs through cancer-derived exosomes binding to TLR2, emphasizing the Toll-like receptors’ role in the crosstalk between inflammation and cancer initiation and progression.
The release of pro-inflammatory cytokines within the TME also recruits neutrophils (the most abundant leukocytes in the immune system) to the TME [32], leading to the generation of reactive oxygen species (ROS) [34][39]. The oxidative stress can lead to single and/or double-strand DNA breaks [40][41], suggesting that exosomes can indirectly increase the genomic instability in the pre-cancer and cancer microenvironment, contributing to cancer initiation and heterogeneity.
In this sense, Abd Elmageed et al. [42] showed that prostate cancer cell-derived exosomes are involved in tumor clonal expansion by reprogramming adipose-derived stem cells via trafficking of oncogenic transcripts (H-ras, K-ras, miR-125b, miR-130b, and miR-155). Supporting these data, Melo et al. [43] demonstrated that exosomes derived from cells and sera of breast cancer patients could promote the formation of tumors from nontumorigenic epithelial cells in a Dicer-dependent manner.
3.2. Cancer-Derived Exosomes Regulate Tumor Promotion and Progression
Although it is clear that cancer-driving mutations are necessary to its initiation, these mutations are not enough to promote its development [44][45].
Thus, cancer development requires sustaining proliferative signals to guarantee the clonal expansion of initiated cells, a step known as cancer promotion. In this sense, two pathways are commonly upregulated in most malignancies: activation of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR [45].
In this sense, several studies have shown that cancer-derived exosomes can provide autocrine, paracrine, and endocrine signals, increasing the proliferation rate of non-cancer and cancer cells [46][47], contributing to both cancer promotion and progression [48][49].
Besides promoting the upregulation of cell-cycle-related genes and increasing the S phase entry, cancer-derived exosomes can also downregulate the expression of cell cycle-arrest-related genes, contributing to the evasion of apoptosis. This is because esophageal adenocarcinoma-derived exosomes and microvesicles could promote the post-transcriptional downregulation of the phosphatase and tensin homolog (PTEN) and the apoptosis-inducing factor 2 (AIFM2) gene in a miR-25- and miR-210-dependent manner [50].
3.3. Cancer-Derived Exosomes Regulate Several Steps of the Metastatic Process
3.3.1. Cancer-Derived Exosomes as a Key Regulator of the Epithelial–Mesenchymal Transition (EMT)
Undoubtedly, metastasis is the most dramatic consequence of cancer, responsible for about 90% of cancer deaths globally [51].
Metastasis is a multistep process, which involves local invasion, intravasation, transport, extravasation, and colonization [52]. These steps require a series of genetic, biochemical, and morphological deregulations that are present in an evolutionarily conserved developmental program known as the epithelial–mesenchymal transition (EMT) [40][53][54][55].
The EMT is a natural process of transdifferentiation of epithelial cells to mesenchymal cells that is crucial for embryogenesis [56][57][58] and re-epithelization in tissue repair [59]. During embryogenesis, the EMT (EMT type I) gives rise to mesoderm (responsible for the formation of muscle, bone, and connective tissues) during gastrulation and neural crest delamination (which results in glial cell, adrenal gland, and epithelial pigmented cell formation) [54][60]. In adult life, the EMT plays a key role in tissue re-epithelization during wound healing (EMT type II) [59][61][62] but, when inappropriately active, such as occurs in carcinogenesis (EMT type III), the EMT causes important disturbances in epithelial tissue homeostasis and integrity, leading to cancer cell spread and metastasis [53][63].
The EMT (type III) is a consequence of cancer progression away from the cancer cells from the stroma, which is responsible for providing nutrients and oxygen support to the cells, creating a hypoxic environment. In addition, the partial reduction in the oxygen pressure leads to the activation of hypoxia-inducible factor 1 alpha (HIF-1α) in both cancer cells and cancer-associated fibroblasts (CAFs) [64][65][66].
HIF-1α nuclear translocation promotes the upregulation and stabilization of Snail and Twist, resulting in cadherin switching, which is characterized by the downregulation of E-cadherin (leading to a loss of intercellular adhesion and consequent activation of the Wnt/β-catenin pathway) and N-cadherin upregulation in cancer cells [67][68][69]. Combined with the F-actin reorganization of invadopodia sites, these actions create sites of transient adhesion that confer cell motility, facilitating the dissemination of cancer cells [53][70].
HIF-1α also acts as a key regulator of metabolic plasticity, promoting genetic and metabolic deregulations [54][71][72]. These deregulations drive the oxidative metabolism to glycolytic metabolism. This process is crucial to guaranteeing the energy supply (ATP) in hypoxic conditions [54]. In addition, glycolytic metabolism increases lactate production, which is generated as a byproduct of glycolysis.
L-Lactate is an important oncometabolite produced by the glycolytic cells within the TME, promoting a metabolic symbiosis between cancer cells and cancer-associated fibroblasts (CAFs) [73]. However, due to its high toxicity, L-lactate is transported out of the cytoplasm of CAFs to the extracellular compartment by a monocarboxylate transporter (MCT4), whose expression is upregulated by HIF-1α [74]. Thus, when released into the TME, the L-lactated CAFs can be uptaken by the MCT1 present in the plasma membrane of glycolytic cancer cells, which acts as a fuel source [75]. This is because cancer cells can oxidize the L-lactate to pyruvate in the mitochondria by lactate dehydrogenase, providing intermediate metabolites to the tricarboxylic acid cycle (TCA) [75][76].
However, the L-lactate exported to the extracellular space promotes the acidification of the TME [75]. The TME’s acidification inhibits the activation and proliferation of CD4+ and CD8+ lymphocytes, natural killer (NK) cells, and dendritic cells (DC) [75] as well as causes the polarization of the macrophages toward the M2 phenotype [75], contributing to immune evasion, which is recognized as a hallmark of cancer [77]. The TME’s acidification also induces the synthesis of metalloproteinases (MMPs) in both cancer and stromal cells, facilitating extracellular matrix (ECM) degradation and, therefore, cancer cell migration and spread [54][78].
Interestingly, studies have demonstrated that activation of HIF-1α by hypoxia increases the secretion of exosomes in both cancer [79][80][81][82] and non-cancer cells within the TME [83][84]. For this reason, hypoxia has been explored to increase the production of mesenchymal stem cell-derived exosomes for novel therapeutic strategies based on cell-free therapy [10][84][85]. This occurs because the hypoxia increases the L-lactate production and, therefore, reduces the pH, increasing the exosome release and uptake, contributing to the crosstalk between cancer and non-cancer cells within the TME [86][87][88].
In this sense, numerous studies have provided evidence that hypoxic cancer-derived exosomes regulate different EMT-related pathways in a miRNA-dependent manner [82][89][90]. In this context, it was reported that the miR-665 identified in hepatocellular carcinoma-derived exosomes can downregulate Hippo signaling through directly targeting tyrosine phosphatase receptor type B (PTPRB) [91], serving as a novel invasive biomarker for this malignancy [92]. This is because the Hippo tumor suppressor signaling pathway is crucial to controlling cell proliferation and apoptosis by inhibiting the oncogenic coactivators Yes-associated protein (YAP)/transcriptional coactivator with the PDZ-binding motif (TAZ) [93][94].
However, considering the plethora of biomolecules, especially miRNAs, delivered by cancer-derived exosomes, the mechanism of action of these vesicles on EMT could not be limited only to the Hippo signaling pathways.
In this sense, Yue et al. [95] showed that exosomal miR-301a, secreted by hypoxic glioblastoma cells, targets transcription elongation factor A like 7 (TCEAL7), leading to the activation of the Wnt/β-catenin signaling pathway, resulting in the expression of the EMT-related transcription factors Snail, Slug, and Twist. Similar results were verified by Nam et al. [96], who demonstrated that miR-301a functions as an oncogene in prostate cancer by directly targeting the p63 tumor suppressor, leading to loss of E-cadherin and EMT.
Thus, it is not surprising that cancer-derived exosomes can regulate different steps of the EMT, including cancer progression [97], dissemination [98][99], ECM remodeling [100][101], stemness [102], and metastasis [103], though different miRNAs.
Interestingly, studies have demonstrated that exosomes derived from cancer-associated macrophages can also regulate stem cells’ dormancy [104] and cell migration and invasion [105], providing evidence that exosomes are also implicated in metastasis.
In this sense, lung cancer cell-derived exosomes (from the A59 and H358 cell lines) alter the transcriptional and bioenergetic signature of M0 macrophages, leading them to an M2 phenotype [106]. However, the M2 macrophage-derived exosomes can transfer miR-21-5p and miR-155-5p to cancer cells, promoting the downregulation of transcription factor Brahma-related gene-1 (BRG1), leading to cell migration and invasion in colon cancer cells [105][107]. Gastric cancer showed similar results; M2 macrophage-derived exosome-mediated apolipoprotein E (ApoE) transfer was found to increase the cancer cell migration in a PI3K/Akt signaling pathway activation-dependent manner [108].
3.3.2. Exosomes in Angiogenesis
Tumor vascularization is crucial to guaranteeing the support of nutrients and meeting oxygen needs to sustain cancer growth. For this reason, the activation of HIF-1α also serves as a signal to induce sustained angiogenesis [64][109]. Once phosphorylated, HIF-1α induces the expression of vascular endothelial growth factor (VEGF) [109][110][111][112]. VEGF binds to VEGF receptors (VEGFRs)-1, -2, and -3, which are expressed on vascular endothelial cells, regulating vessel formation through endothelial cell migration [113][114].
In this context, studies have demonstrated that cancer-derived exosomes act as a key regulator of angiogenesis [115][116]. This is because exosomes derived from cancer cells can stimulate endothelial cell migration and tube formation independently of uptake [117]. This response is mediated by the 189-amino-acid heparin-bound isoform of VEGF, which, unlike other common isoforms of VEGF, is preferentially enriched on the exosome surface [117].
However, cancer-derived exosomes can also promote angiogenesis in an uptake-dependent manner. In this sense, Li et al. [118] showed that hepatocellular carcinoma-derived exosomes transporting lysyl oxidase-like 4 (LOXL4) induce angiogenesis. In another study, Zhang et al. [119] demonstrated that ovarian cancer-derived exosomes expressing prokineticin receptor 1 (PKR1) promote angiogenesis by promoting the migration and tube formation of HUVEC cells. Similar results were also described by Umezu et al. [120], who demonstrated that hypoxia increases the production of multiple myeloma cell-derived exosomes transporting miR-135b, which can bind to factor-inhibiting hypoxia-inducible factor 1 (FIH-1) in endothelial cells, enhancing the formation of endothelial tubes. In another study, Zeng et al. [121] showed that colorectal cancer-derived exosomes drive miR-25-3p to endothelial cells, targeting Kruppel-like factors 1 and 4 (KLF2 and KF4, respectively) and promoting vascular permeability and angiogenesis.
3.3.3. Cancer-Derived Exosomes Contribute to Pre-Metastatic Niche (PMN) Formation
Angiogenesis contributes to both cancer cell and cancer-derived exosome dissemination. However, the outcome of cancer metastasis depends on the interactions between metastatic cells and the host microenvironment [122]. These interactions between the cancer cells (“seeds”) and the host microenvironment (“soils”) were first discovered by the English surgeon Stephen Paget in 1889 [122]. About 40 years later (in 1928), James Ewing postulated that metastasis is determined by a mechanism associated with hemodynamic factors of the vascular system [123]. In a complementary hypothesis postulated in the 1970s, Isaiah Fidler demonstrated that, although the mechanical properties of blood flow are important, metastatic colonization only occurs at certain organ sites (organotropism) [123]. Fidler’s theory was supported by additional discoveries, which revealed that tumors induce the formation of microenvironments in distant organs, facilitating the survival and outgrowth of cancer cells before they arrived at these sites [123][124][125][126]. These predetermined microenvironments are termed ‘pre-metastatic niches’ (PMNs) [127].
In the context of the “seed and soil” theory (Paget’s theory), the exosomes are similar to fertilizers, which can make barren land fertile and facilitate the colonization of cancer cells [127][128][129][130]. This occurs because exosomes exhibit adhesion molecules on their surface, particularly integrins (ITGs), which bind to the ECM and organ-specific PMN receptors [128]. Supporting this theory, in a study evaluating the biodistribution of exosomes from different cancer cell lines, Hoshino et al. [131] provided evidence that cancer-derived exosomes are preferentially uptaken by tissues commonly recognized as metastatic sites. The authors also demonstrated that this site-specific biodistribution is associated with high expression levels of integrins (ITGα6, ITGβ4, and ITGβ1 for lung tropism; ITGβ5 and ITGαv for liver tropism; and ITGβ3 for brain tropism) [131], reinforcing the view that the integrins involved in PMN formation.
Cumulative studies have provided evidence that the local inflammatory microenvironment drives the formation of PMNs as revisited by Guo et al. [127]. In this sense, the exosomes play a key role in the metastatic process, inducing immune suppression in the PMN. This is because cancer cells release exosomes carrying programmed death-ligand 1 (PD-L1) [127]. When PD-L1 binds to programmed death receptor 1 (PD-1), which is mainly expressed on macrophages and activated T or B cells, it provides an inhibitory signal, inducing T cell apoptosis and/or inhibiting T cell activation and proliferation [132]. Thus, PD-L1/PD-1 binding allows the exosomes to circulate through the bloodstream without being recognized by immune cells [127][133][134].
In addition, cancer-derived exosomes contain many immunomodulatory molecules that can impair the immune cell function, resulting in an immunosuppressive pre-metastatic microenvironment [127]. These molecules can induce natural killer (NK) cell dysfunction, inhibit antigen-presenting cells, block T cell activation, and enhance apoptosis [135][136].
However, the effects of cancer-derived exosomes in PMN formation are not limited to immune suppression. Studies have demonstrated that exosomes released from hypoxic tumors increase angiogenesis and vascular permeability in the PMN by carrying different miRNAs, such as miR-105 and miR-25-3p, which can disrupt the vascular endothelial barrier by targeting specific gene products [130][131][137].
3.3.4. Exosomes in Cancer Stem Cell (CSC) Formation
Cancer stem cells (CSCs), also known as tumor-initiating cells (TICs), are a subset of cancer cells that share various features with stem cells, including the ability to self-renew and differentiation into the heterogeneous lineages of cancer cells, producing a variety of tumor cell subpopulations [138][139][140][141]. In addition, these cells can induce cell cycle arrest (quiescent state), conferring chemo- and radio-resistance. This is because many common chemotherapeutic agents target the proliferating cells to lead to their apoptosis [139]. Furthermore, CSCs overexpress ATP-binding cassette (ABC) transporters, increasing chemotherapeutics’ efflux [142][143][144]. In addition, by exhibiting a high capability to repair DNA damage, the CSCs are resistant to radiation therapy (RT) [145][146]. Thus, although the origin of CSCs remains incompletely understood [147], it is clear that these cells are currently involved in therapeutic resistance [148].
Cumulative evidence has shown that genomic instability contributes to CSC formation and accelerates the development of many genetically variable cancer stem cells, increasing the intratumor heterogeneity [53][149][150][151][152].
However, recent studies have provided evidence that cancer-derived exosomes mediate crosstalk between the EMT and cancer stem cell (CSC) formation, acting as a key regulator of cell plasticity [138].
In this sense, numerous studies have shown that cancer-derived exosomes mediate the instability of cadherins (which was verified during the EMT) in recipient cells by transferring oncogenic microRNAs and long non-coding RNAs (lncRNAs) as revisited by Wang et al. [153].
The loss of E-cadherin, mediated by these non-coding RNAs [153], promotes β-catenin release into the cytoplasm [154]. Once translocated to the nucleus, β-catenin downregulates not only cell-junction-related genes (E-cadherin and claudin-7) [53][155] but also upregulates stemness-related genes, facilitating the formation of CSCs [156][157][158].
In addition, studies have also demonstrated that cancer-derived exosomes mediate drug resistance in several malignancies, which is considered a major impediment in medical oncology [159].
Basically, there are two main types of resistance in cancer: (i) inherent resistance, where insensitivity already exists before treatment; and (ii) acquired resistance, which subsequently appears following the initial positive response [159]. Interestingly, studies have demonstrated that cancer-derived exosomes mediate the acquired resistance by transferring microRNAs as revised by Bach et al. [159].
References
- Golchin, A.; Hosseinzadeh, S.; Ardeshirylajimi, A. The exosomes released from different cell types and their effects in wound healing. J. Cell. Biochem. 2018, 119, 5043–5052.
- Baixauli, F.; LÃ3pez-Otà n, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403.
- Baek, G.; Choi, H.; Kim, Y.; Lee, H.C.; Choi, C. Mesenchymal stem cell-derived extracellular vesicles as therapeutics and as a drug delivery platform. Stem Cells Transl. Med. 2019, 8, 880–886.
- Bu, H.; He, D.; He, X.; Wang, K. Exosomes: Isolation, Analysis, and Applications in Cancer Detection and Therapy. ChemBioChem 2019, 20, 451–461.
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular vesicles: Unique intercellular delivery vehicles. Trends Cell Biol. 2017, 27, 172–188.
- Antounians, L.; Tzanetakis, A.; Pellerito, O.; Catania, V.D.; Sulistyo, A.; Montalva, L.; McVey, M.J.; Zani, A. The regenerative potential of amniotic fluid stem cell extracellular vesicles: Lessons learned by comparing different isolation techniques. Sci. Rep. 2019, 9, 1837.
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579.
- Cheng, L.; Zhang, K.; Wu, S.; Cui, M.; Xu, T. Focus on mesenchymal stem cell-derived exosomes: Opportunities and challenges in cell-free therapy. Stem Cells Int. 2017, 2017, 6305295.
- Exosomes, E.; Vivo, I.; Verweij, F.J.; Revenu, C.; Arras, G.; Del Bene, F.; Van Niel, G.; Verweij, F.J.; Revenu, C.; Arras, G.; et al. Live Tracking of Inter-organ Communication by Resource Live Tracking of Inter-organ Communication by Endogenous Exosomes In Vivo. Dev. Cell 2019, 48, 573–589.
- Araldi, R.P.; D’Amelio, F.; Vigerelli, H.; de Melo, T.C.; Kerkis, I. Stem cell-derived exosomes as therapeutic approach for neurodegenerative Ddsorders: From biology to biotechnology. Cells 2020, 9, 2663.
- Anand, S.; Samuel, M.; Kumar, S.; Mathivanan, S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 140203.
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19.
- Rezaie, J.; Ajezi, S.; Avci, Ç.B.; Karimipour, M.; Geranmayeh, M.H.; Nourazarian, A.; Sokullu, E.; Rezabakhsh, A.; Rahbarghazi, R. Exosomes and their application in biomedical field: Difficulties and advantages. Mol. Neurobiol. 2018, 55, 3372–3393.
- Villarroya-Beltri, C.; Baixauli, F.; Gutiérrez-Vázquez, C.; Sánchez-Madrid, F.; Mittelbrunn, M. Sorting it out: Regulation of exosome loading. Semin. Cancer Biol. 2014, 28, 3–13.
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228.
- Wei, D.; Zhan, W.; Gao, Y.; Huang, L.; Gong, R.; Wang, W.; Zhang, R.; Wu, Y.; Gao, S.; Kang, T. RAB31 marks and controls an ESCRT-independent exosome pathway. Cell Res. 2021, 31, 157–177.
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937.
- Airola, M.V.; Hannun, Y.A. Sphingolipid metabolism and neutral sphingomyelinases. Handb. Exp. Pharmacol. 2013, 215, 57–76.
- Al-Sowayan, B.S.; Al-Shareeda, A.T.; Alrfaei, B.M. Cancer stem cell-exosomes, unexposed player in tumorigenicity. Front. Pharmacol. 2020, 11, 384.
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360.
- Jaiswal, R.; Sedger, L.M. Intercellular vesicular transfer by exosomes, microparticles and oncosomes-implications for cancer biology and treatments. Front. Oncol. 2019, 9, 125.
- Meehan, B.; Rak, J.; Di Vizio, D. Oncosomes—large and small: What are they, where they came from? J. Extracell. Vesicles 2016, 5, 33109.
- Minciacchi, V.R.; You, S.; Spinelli, C.; Morley, S.; Zandian, M.; Aspuria, P.-J.; Cavallini, L.; Ciardiello, C.; Sobreiro, M.R.; Morello, M.; et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget 2015, 6, 11327–11341.
- Ma, L.; Li, Y.; Peng, J.; Wu, D.; Zhao, X.; Cui, Y.; Chen, L.; Yan, X.; Du, Y.; Yu, L. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015, 25, 24–38.
- Hanahan, D.; Weinberg, R. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 25–50.
- Liu, S.-L.; Sun, P.; Li, Y.; Liu, S.-S.; Lu, Y. Exosomes as critical mediators of cell-to-cell communication in cancer pathogenesis and their potential clinical application. Transl. Cancer Res. 2019, 8, 298–311.
- Stefanius, K.; Servage, K.; de Souza Santos, M.; Gray, H.F.; Toombs, J.E.; Chimalapati, S.; Kim, M.S.; Malladi, V.S.; Brekken, R.; Orth, K. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. Elife 2019, 8, e40226.
- Chan, B.D.; Wong, W.; Lee, M.M.; Cho, W.C.; Yee, B.K.; Kwan, Y.W.; Tai, W.C. Exosomes in inflammation and inflammatory disease. Proteomics 2019, 19, 1800149.
- Weber, D. Inflammation and cancer: Tumor initiation, progression and metastasis, and Chinese botanical medicines. J. Chin. Integr. Med. 2010, 8, 1006–1013.
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41.
- Singel, K.L.; Segal, B.H. Neutrophils in the tumor microenvironment: Trying to heal the wound that cannot heal. Immunol. Rev. 2016, 273, 329–343.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899.
- Stefanius, K.; Servage, K.; Orth, K. Exosomes in cancer development. Curr. Opin. Genet. Dev. 2021, 66, 83–92.
- Othman, N.; Jamal, R.; Abu, N. Cancer-derived exosomes as effectors of key Inflammation-related players. Front. Immunol. 2019, 10, 2103.
- Bretz, N.P.; Ridinger, J.; Rupp, A.-K.; Rimbach, K.; Keller, S.; Rupp, C.; Marmé, F.; Umansky, L.; Umansky, V.; Eigenbrod, T.; et al. Body Fluid Exosomes Promote Secretion of Inflammatory Cytokines in Monocytic Cells via Toll-like Receptor Signaling. J. Biol. Chem. 2013, 288, 36691–36702.
- Chow, A.; Zhou, W.; Liu, L.; Fong, M.Y.; Champer, J.; Van Haute, D.; Chin, A.R.; Ren, X.; Gugiu, B.G.; Meng, Z.; et al. Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κB. Sci. Rep. 2015, 4, 5750.
- Wu, L.; Zhang, X.; Zhang, B.; Shi, H.; Yuan, X.; Sun, Y.; Pan, Z.; Qian, H.; Xu, W. Exosomes derived from gastric cancer cells activate NF-κB pathway in macrophages to promote cancer progression. Tumor Biol. 2016, 37, 12169–12180.
- Butin-Israeli, V.; Bui, T.M.; Wiesolek, H.L.; Mascarenhas, L.; Lee, J.J.; Mehl, L.C.; Knutson, K.R.; Adam, S.A.; Goldman, R.D.; Beyder, A.; et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Investig. 2019, 129, 712–726.
- De-Sá-Júnio, P.; Câmara, D.; Porcacchia, A.; Fonseca, P.; Jorge, S.; Araldi, R.; Ferreira, A. The roles of ROS in cancer heterogeneity and therapy. Oxid. Med. Cell. Longev. 2017, 2017, 2467940.
- Araldi, R.; Mazzuchelli-de-souza, J.; Modolo, D.; Souza, E.; Melo, T.; Spadacci-Morena, D.; Magnelli, R.; Rocha, M.; De-Sá-Júnior, P.; Carvalho, R.; et al. Mutagenic potential of Bos taurus papillomavirus type 1 E6 recombinant protein: First description. Biomed. Res. Int. 2015, 2015, 806361.
- Abd Elmageed, Z.Y.; Yang, Y.; Thomas, R.; Ranjan, M.; Mondal, D.; Moroz, K.; Fang, Z.; Rezk, B.M.; Moparty, K.; Sikka, S.C.; et al. Neoplastic reprogramming of patient-derived adipose stem cells by prostate cancer cell-associated exosomes. Stem Cells 2014, 32, 983–997.
- Melo, S.A.; Sugimoto, H.; Connell, J.T.O.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721.
- Adjiri, A. DNA mutations may not be the cause of cancer. Oncol. Ther. 2017, 5, 85–101.
- Zhang, D.; Huo, D.; Xie, H.; Wu, L.; Zhang, J.; Liu, L.; Jin, Q.; Chen, X. CHG: A systematically integrated database of cancer hallmark. Front Genet. 2020, 11, 29.
- Koga, K.; Matsumoto, K.; Akiyoshi, T.; Kubo, M.; Yamanaka, N.; Tasaki, A.; Nakashima, H.; Nakamura, M.; Kuroki, S.; Tanaka, M.; et al. Purification, characterization and biological significance of tumor-derived exosomes. Anticancer Res. 2005, 25, 3703–3707.
- Yang, L.; Wu, X.-H.; Wang, D.; Luo, C.-L.; Chen, L.-X. Bladder cancer cell-derived exosomes inhibit tumor cell apoptosis and induce cell proliferation in vitro. Mol. Med. Rep. 2013, 8, 1272–1278.
- Osaki, M.; Okada, F. Exosomes and their role in cancer progression. Yonago Acta Med. 2019, 62, 182–190.
- Yu, S.; Cao, H.; Shen, B.; Feng, J. Tumor-derived exosomes in cancer progression and treatment failure. Oncotarget 2015, 6, 37151–37168.
- Ke, X.; Yan, R.; Sun, Z.; Cheng, Y.; Meltzer, A.; Lu, N.; Shu, X.; Wang, Z.; Huang, B.; Liu, X.; et al. Esophageal adenocarcinoma–derived extracellular vesicle microRNAs induce a neoplastic phenotype in gastric organoids. Neoplasia 2017, 19, 941–949.
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564.
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206.
- Araldi, R.P.; de Melo, T.C.; Levy, D.; de Souza, D.M.; Maurício, B.; Colozza-Gama, G.A.; Bydlowski, S.P.; Peng, H.; Rauscher, F.J.; Cerutti, J.M. LIMD2 regulates key steps of metastasis cascade in papillary thyroid cancer cells via MAPK crosstalk. Cells 2020, 9, 2522.
- Araldi, R.; Módolo, D.; De-Sá-Júnior, P.; Consonni, S.; Carvalho, R.; Roperto, F.; Beçak, W.; Stocco, R. Genetics and metabolic deregulation following cancer initiation: A world to explore. Biomed. Pharmacother. 2016, 82, 449–458.
- Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 395–412.
- Cichon, M.; Nelson, C.; Radisky, D. Regulation of epithelial-mesenchymal transition in breast cancer cells by cell contact and adhesion. Cancer Inform. 2015, 14, CIN–S18965.
- Lee, K.; Nelson, C. New Insights into the Regulation of Epithelial-Mesenchymal Transition and Tissue Fibrosis, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 294, ISBN 9780123943057.
- Tian, Y.; Xie, Q.; He, J.; Luo, X.; Zhou, T.; Liu, Y.; Huang, Z.; Tian, Y.; Sun, D.; Yao, K. Radioactive 125 I seeds inhibit cell growth and epithelial-mesenchymal transition in human glioblastoma multiforme via a ROS-mediated signaling pathway. BMC Cancer 2015, 15, 1.
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.-P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor mecrosis factor-α through bone morphogenic protein-2. Am. J. Pathol. 2010, 176, 2247–2258.
- Radisky, D.; LaBarge, M. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2008, 2, 511–512.
- Haensel, D.; Dai, X. Epithelial-to-mesenchymal transition in cutaneous wound healing: Where we are and where we are heading. Dev. Dyn. 2018, 247, 473–480.
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506.
- Wever, O.; Pauwels, P.; De Craene, B.; Sabbah, M.; Emami, S.; Redeuilh, G.; Gespach, C.; Bracke, M.; Berx, G. Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front. Histochem. Cell Biol. 2008, 130, 481–494.
- Fang, J.; Gillies, R.; Gatenby, R. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin. Cancer Biol. 2008, 18, 330–337.
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target. Ther. 2020, 5, 242.
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10.
- Zhang, P.; Liu, Y.; Feng, Y.; Gao, S. SNAIL gene inhibited by hypoxia-inducible factor 1α (HIF-1α) in epithelial ovarian cancer. Int. J. Immunopathol. Pharmacol. 2016, 29, 364–375.
- Lv, L.; Yuan, J.; Huang, T.; Zhang, C.; Zhu, Z.; Wang, L.; Jiang, G.; Zeng, F. Stabilization of Snail by HIF-1α and TNF-α is required for hypoxia-induced invasion in prostate cancer PC3 cells. Mol. Biol. Rep. 2014, 41, 4573–4582.
- Sun, S.; Ning, X.; Zhang, Y.; Lu, Y.; Nie, Y.; Han, S.; Liu, L.; Du, R.; Xia, L.; He, L.; et al. Hypoxia-inducible factor-1α induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 2009, 75, 1278–1287.
- Araldi, R.; Lima, T.; Módolo, D.; Mazzuchelli-de-Souza, J.; Magnelli, R.; Maurício, B.; Spadacci-Morena, D.; De-Sá-Júnior, P.; Carvalho, R.; Beçak, W.; et al. Analysis of stem-cell and migratory phenotype in primary cultures derived from BPV-infected benign and malignant neoplasms. J. Cancer Res. Ther. Oncol. 2017, 5, 1–13.
- Yang, S.; Zhang, Z.; Hao, Y.; Zhao, Y.; Qian, F.; Shi, Y.; Li, P.; Liu, C.; Yu, P. HIF-1α induces the epithelial-mesenchymal transition in gastric cancer stem cells through the Snail pathway. Oncotarget 2017, 8, 9535–9545.
- Hapke, R.Y.; Haake, S.M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020, 487, 10–20.
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66.
- Kim, H.K.; Lee, I.; Bang, H.; Kim, H.C.; Lee, W.Y.; Yun, S.H.; Lee, J.; Lee, S.J.; Park, Y.S.; Kim, K.-M.; et al. MCT4 expression is a potential therapeutic target in colorectal cancer with peritoneal carcinomatosis. Mol. Cancer Ther. 2018, 17, 838–848.
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143.
- Tomasetti, M.; Lee, W.; Santarelli, L.; Neuzil, J. Exosome-derived microRNAs in cancer metabolism: Possible implications in cancer diagnostics and therapy. Exp. Mol. Med. 2017, 49, e285.
- Mortezaee, K. Immune scape: A critical hallmark in solid tumors. Life iSci. 2020, 258, 118110.
- Gonzalez-Avila, G.; Sommer, B.; García-Hernández, A.A.; Ramos, C. Matrix Metalloproteinases’ Role in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 97–131.
- Panigrahi, G.K.; Praharaj, P.P.; Peak, T.C.; Long, J.; Singh, R.; Rhim, J.S.; Abd Elmageed, Z.Y.; Deep, G. Hypoxia-induced exosome secretion promotes survival of African-American and Caucasian prostate cancer cells. Sci. Rep. 2018, 8, 3853.
- Meng, W.; Hao, Y.; He, C.; Li, L.; Zhu, G. Exosome-orchestrated hypoxic tumor microenvironment. Mol. Cancer 2019, 18, 57.
- Kore, R.A.; Edmondson, J.L.; Jenkins, S.V.; Jamshidi-Parsian, A.; Dings, R.P.M.; Reyna, N.S.; Griffin, R.J. Hypoxia-derived exosomes induce putative altered pathways in biosynthesis and ion regulatory channels in glioblastoma cells. Biochem. Biophys. Rep. 2018, 14, 104–113.
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421.
- Zhang, W.; Zhou, X.; Yao, Q.; Liu, Y.; Zhang, H.; Dong, Z. HIF-1-mediated production of exosomes during hypoxia is protective in renal tubular cells. Am. J. Physiol.-Ren. Physiol. 2017, 313, F906–F913.
- Liu, W.; Li, L.; Rong, Y.; Qian, D.; Chen, J.; Zhou, Z.; Luo, Y.; Jiang, D.; Cheng, L.; Zhao, S.; et al. Hypoxic mesenchymal stem cell-derived exosomes promote bone fracture healing by the transfer of miR-126. Acta Biomater. 2020, 103, 196–212.
- Cheng, H.; Chang, S.; Xu, R.; Chen, L.; Song, X.; Wu, J.; Qian, J.; Zou, Y.; Ma, J. Hypoxia-challenged MSC-derived exosomes deliver miR-210 to attenuate post-infarction cardiac apoptosis. Stem Cell Res. Ther. 2020, 11, 224.
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2016, 38, 119–133.
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222.
- Boussadia, Z.; Zanetti, C.; Parolini, I. Role of microenvironmental acidity and tumor exosomes in cancer immunomodulation. Transl. Cancer Res. 2020, 9, 5775–5786.
- Park, J.E.; Tan, H.S.; Datta, A.; Lai, R.C.; Zhang, H.; Meng, W.; Lim, S.K.; Sze, S.K. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol. Cell. Proteom. 2010, 9, 1085–1099.
- Svensson, K.J.; Kucharzewska, P.; Christianson, H.C.; Sköld, S.; Löfstedt, T.; Johansson, M.C.; Mörgelin, M.; Bengzon, J.; Ruf, W.; Belting, M. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13147–13152.
- Hu, Y.; Yang, C.; Yang, S.; Cheng, F.; Rao, J.; Wang, X. miR-665 promotes hepatocellular carcinoma cell migration, invasion, and proliferation by decreasing Hippo signaling through targeting PTPRB. Cell Death Dis. 2018, 9, 954.
- Qu, Z.; Wu, J.; Wu, J.; Ji, A.; Qiang, G.; Jiang, Y.; Jiang, C.; Ding, Y. Exosomal miR-665 as a novel minimally invasive biomarker for hepatocellular carcinoma diagnosis and prognosis. Oncotarget 2017, 8, 80666–80678.
- Kim, H.; Lee, S.; Shin, E.; Seong, K.M.; Jin, Y.W.; Youn, H.; Youn, B. The emerging roles of exosomes as EMT regulators in cancer. Cells 2020, 9, 861.
- Boopathy, G.T.K.; Hong, W. Role of Hippo pathway-YAP/TAZ signaling in angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49.
- Yue, X.; Lan, F.; Xia, T. Hypoxic Glioma Cell-Secreted Exosomal miR-301a Activates Wnt/β-catenin Signaling and Promotes Radiation Resistance by Targeting TCEAL7. Mol. Ther. 2019, 27, 1939–1949.
- Nam, R.K.; Benatar, T.; Wallis, C.J.D.; Amemiya, Y.; Yang, W.; Garbens, A.; Naeim, M.; Sherman, C.; Sugar, L.; Seth, A. MiR-301a regulates E-cadherin expression and is predictive of prostate cancer recurrence. Prostate 2016, 76, 869–884.
- Hu, X.; Mu, Y.; Liu, J.; Mu, X.; Gao, F.; Chen, L.; Wu, H.; Wu, H.; Liu, W.; Zhao, Y. Exosomes derived from hypoxic colorectal cancer cells transfer miR-410-3p to regulate tumor progression. J. Cancer 2020, 11, 4724–4735.
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-Secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515.
- Hoshino, D.; Kirkbride, K.C.; Costello, K.; Clark, E.S.; Sinha, S.; Grega-Larson, N.; Tyska, M.J.; Weaver, A.M. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 2013, 5, 1159–1168.
- Mu, W.; Rana, S.; Zöller, M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia 2013, 15, 875–887.
- Ge, R.; Tan, E.; Sharghi-Namini, S.; Asada, H.H. Exosomes in cancer microenvironment and beyond: Have we overlooked these extracellular messengers? Cancer Microenviron. 2012, 5, 323–332.
- Kling, M.J.; Chaturvedi, N.K.; Kesherwani, V.; Coulter, D.W.; McGuire, T.R.; Sharp, J.G.; Joshi, S.S. Exosomes secreted under hypoxia enhance stemness in Ewing’s sarcoma through miR-210 delivery. Oncotarget 2020, 11, 3633–3645.
- Mao, Y.; Wang, Y.; Dong, L.; Zhang, Y.; Zhang, Y.; Wang, C.; Zhang, Q.; Yang, S.; Cao, L.; Zhang, X.; et al. Hypoxic exosomes facilitate angiogenesis and metastasis in esophageal squamous cell carcinoma through altering the phenotype and transcriptome of endothelial cells. J. Exp. Clin. Cancer Res. 2019, 38, 389.
- Walker, N.D.; Elias, M.; Guiro, K.; Bhatia, R.; Greco, S.J.; Bryan, M.; Gergues, M.; Sandiford, O.A.; Ponzio, N.M.; Leibovich, S.J.; et al. Exosomes from differentially activated macrophages influence dormancy or resurgence of breast cancer cells within bone marrow stroma. Cell Death Dis. 2019, 10, 59.
- Lan, J.; Sun, L.; Xu, F.; Liu, L.; Hu, F.; Song, D.; Hou, Z.; Wu, W.; Luo, X.; Wang, J.; et al. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. 2019, 79, 146–158.
- Pritchard, A.; Tousif, S.; Wang, Y.; Hough, K.; Khan, S.; Strenkowski, J.; Chacko, B.K.; Darley-Usmar, V.M.; Deshane, J.S. Lung tumor cell-derived exosomes promote M2 macrophage polarization. Cells 2020, 9, 1303.
- Ma, Y.-S.; Wu, T.-M.; Ling, C.-C.; Yu, F.; Zhang, J.; Cao, P.-S.; Gu, L.-P.; Wang, H.-M.; Xu, H.; Li, L.; et al. M2 macrophage-derived exosomal microRNA-155-5p promotes the immune escape of colon cancer by downregulating ZC3H12B. Mol. Ther. Oncolytics 2021, 20, 484–498.
- Zheng, P.; Luo, Q.; Wang, W.; Li, J.; Wang, T.; Wang, P.; Chen, L.; Zhang, P.; Chen, H.; Liu, Y.; et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional Apolipoprotein E. Cell Death Dis. 2018, 9, 434.
- Shi, Y.-H.; Fang, W.-G. Hypoxia-inducible factor-1 in tumour angiogenesis. World J. Gastroenterol. 2004, 10, 1082.
- Hashimoto, T.; Shibasaki, F. Hypoxia-inducible factor as an angiogenic master switch. Front. Pediatr. 2015, 3, 33.
- Harada, H.; Kizaka-Kondoh, S.; Li, G.; Itasaka, S.; Shibuya, K.; Inoue, M.; Hiraoka, M. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene 2007, 26, 7508–7516.
- Lin, C.; McGough, R.; Aswad, B.; Block, J.A.; Terek, R. Hypoxia induces HIF-1Œ± and VEGF expression in chondrosarcoma cells and chondrocytes. J. Orthop. Res. 2004, 22, 1175–1181.
- Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105.
- Cébe-Suarez, S.; Zehnder-Fjällman, A.; Ballmer-Hofer, K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell. Mol. Life Sci. 2006, 63, 601–615.
- Aslan, C.; Maralbashi, S.; Salari, F.; Kahroba, H.; Sigaroodi, F.; Kazemi, T.; Kharaziha, P. Tumor-derived exosomes: Implication in angiogenesis and antiangiogenesis cancer therapy. J. Cell. Physiol. 2019, 234, 16885–16903.
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476.
- Ko, S.Y.; Lee, W.; Kenny, H.A.; Dang, L.H.; Ellis, L.M.; Jonasch, E.; Lengyel, E.; Naora, H. Cancer-derived small extracellular vesicles promote angiogenesis by heparin-bound, bevacizumab-insensitive VEGF, independent of vesicle uptake. Commun. Biol. 2019, 2, 386.
- Li, R.; Wang, Y.; Zhang, X.; Feng, M.; Ma, J.; Li, J.; Yang, X.; Fang, F.; Xia, Q.; Zhang, Z.; et al. Exosome-mediated secretion of LOXL4 promotes hepatocellular carcinoma cell invasion and metastasis. Mol. Cancer 2019, 18, 18.
- Zhang, X.; Sheng, Y.; Li, B.; Wang, Q.; Liu, X.; Han, J. Ovarian cancer derived PKR1 positive exosomes promote angiogenesis by promoting migration and tube formation in vitro. Cell Biochem. Funct. 2021, 39, 308–316.
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757.
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395.
- Ribatti, D.; Mangialardi, G.; Vacca, A. Stephen Paget and the ‘seed and soil’ theory of metastatic dissemination. Clin. Exp. Med. 2006, 6, 145–149.
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176.
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293.
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827.
- Sleeman, J.P. The lymph node pre-metastatic niche. J. Mol. Med. 2015, 93, 1173–1184.
- Guo, Y.; Ji, X.; Liu, J.; Fan, D.; Zhou, Q.; Chen, C.; Wang, W.; Wang, G.; Wang, H.; Yuan, W.; et al. Effects of exosomes on pre-metastatic niche formation in tumors. Mol. Cancer 2019, 18, 39.
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317.
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826.
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891.
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335.
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704.
- Theodoraki, M.-N.; Yerneni, S.S.; Hoffmann, T.K.; Gooding, W.E.; Whiteside, T.L. Clinical significance of PD-L1 + exosomes in plasma of head and neck cancer patients. Clin. Cancer Res. 2018, 24, 896–905.
- Hong, C.-S.; Sharma, P.; Yerneni, S.S.; Simms, P.; Jackson, E.K.; Whiteside, T.L.; Boyiadzis, M. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci. Rep. 2017, 7, 14684.
- Ludwig, S.; Floros, T.; Theodoraki, M.-N.; Hong, C.-S.; Jackson, E.K.; Lang, S.; Whiteside, T.L. Suppression of lymphocyte functions by plasma exosomes correlates with disease activity in patients with head and neck cancer. Clin. Cancer Res. 2017, 23, 4843–4854.
- Czernek, L.; Düchler, M. Functions of cancer-derived extracellular vesicles in immunosuppression. Arch. Immunol. Ther. Exp. 2017, 65, 311–323.
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringner, M.; Morgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317.
- Xu, J.; Liao, K.; Zhou, W. Exosomes regulate the transformation of cancer cells in cancer stem cell homeostasis. Stem Cells Int. 2018, 2018, 4837370.
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, 5416923.
- Clarke, M.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115.
- Islam, F.; Qiao, B.; Smith, R.A.; Gopalan, V.; Lam, A.K.Y. Cancer stem cell: Fundamental experimental pathological concepts and updates. Exp. Mol. Pathol. 2015, 98, 184–191.
- Begicevic, R.-R.; Falasca, M. ABC transporters in cancer stem cells: Beyond chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362.
- Han, S.; Kim, J.; Kim, M.; Kim, J.; Lee, K.; Kim, B.; Oh, H.; Kim, D.; Kang, S.; Kim, H.; et al. Prognostic implication of ABC transporters and cancer stem cell markers in patients with stage III colon cancer receiving adjuvant FOLFOX-4 chemotherapy. Oncol. Lett. 2019, 17, 5572–5580.
- Abbasifarid, E.; Sajjadi-Jazi, S.M.; Beheshtian, M.; Samimi, H.; Larijani, B.; Haghpanah, V. The role of ATPbBinding cassette transporters in the chemoresistance of anaplastic thyroid cancer: A systematic review. Endocrinology 2019, 160, 2015–2023.
- Olivares-Urbano, M.A.; Griñán-Lisón, C.; Marchal, J.A.; Núñez, M.I. CSC radioresistance: A therapeutic challenge to improve radiotherapy effectiveness in cancer. Cells 2020, 9, 1651.
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.-I.; Ganswindt, U. The role of cancer stem cells in radiation resistance. Front. Oncol. 2020, 10, 164.
- Nimmakayala, R.K.; Batra, S.K.; Ponnusamy, M.P. Unraveling the journey of cancer stem cells from origin to metastasis. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 50–63.
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125.
- Lagasse, E. Cancer stem cells with genetic instability: The best vehicle with the best engine for cancer. Gene Ther. 2008, 15, 136–142.
- Varga, J.; Oliveira, T.; Greten, F. The architect who never sleeps: Tumor-induced plasticity. FEBS Lett. 2014, 588, 2422–2427.
- Shipitsin, M.; Polyak, K. The cancer stem cell hypothesis: In search of definitions, markers, and relevance. Lab. Investig. 2008, 88, 459–463.
- Li, L.; Borodyansky, L.; Yang, Y. Genomic instability en route to and from cancer stem cells. Cell Cycle 2009, 8, 1000–1002.
- Wang, B.; Tan, Z.; Guan, F. Tumor-derived exosomes mediate the instability of cadherins and promote tumor progression. Int. J. Mol. Sci. 2019, 20, 3652.
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-cadherin/β -catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 1–6.
- Kim, W.K.; Kwon, Y.; Jang, M.; Park, M.; Kim, J.; Cho, S.; Jang, D.G.; Lee, W.-B.; Jung, S.H.; Choi, H.J.; et al. β-catenin activation down-regulates cell-cell junction-related genes and induces epithelial-to-mesenchymal transition in colorectal cancers. Sci. Rep. 2019, 9, 18440.
- Mavila, N.; Thundimadathil, J. The emerging roles of cancer stem cells and Wnt/Beta-catenin signaling in hepatoblastoma. Cancers 2019, 11, 1406.
- Tanaka, H.; Kawaguchi, M.; Shoda, S.; Miyoshi, T.; Iwasaki, R.; Hyodo, F.; Mori, T.; Hara, A.; Tomita, H.; Matsuo, M. Nuclear accumulation of β-catenin in cancer stem Cell radioresistance and stemness in human colon cancer. Anticancer Res. 2019, 39, 6575–6583.
- Pandit, H.; Li, Y.; Li, X.; Zhang, W.; Li, S.; Martin, R.C.G. Enrichment of cancer stem cells via β-catenin contributing to the tumorigenesis of hepatocellular carcinoma. BMC Cancer 2018, 18, 783.
- Bach, D.-H.; Hong, J.-Y.; Park, H.J.; Lee, S.K. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int. J. Cancer 2017, 141, 220–230.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
22 Nov 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No