Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ferdinando Nicoletti | + 2214 word(s) | 2214 | 2021-11-10 05:16:41 | | | |
| 2 | Peter Tang | Meta information modification | 2214 | 2021-11-19 02:38:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nicoletti, F. Macrophage Migration Inhibitory Factor Family in Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/16148 (accessed on 25 July 2026).
Nicoletti F. Macrophage Migration Inhibitory Factor Family in Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/16148. Accessed July 25, 2026.
Nicoletti, Ferdinando. "Macrophage Migration Inhibitory Factor Family in Cancer" Encyclopedia, https://encyclopedia.pub/entry/16148 (accessed July 25, 2026).
Nicoletti, F. (2021, November 18). Macrophage Migration Inhibitory Factor Family in Cancer. In Encyclopedia. https://encyclopedia.pub/entry/16148
Nicoletti, Ferdinando. "Macrophage Migration Inhibitory Factor Family in Cancer." Encyclopedia. Web. 18 November, 2021.
Copy Citation
Macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine that plays a key role in several diseases, including cancer. Preclinical and clinical studies in NB patients convergently indicate that MIF exerts pro-tumorigenic properties in NB. MIF is upregulated in NB tumor tissues and cell lines and it contributes to NB aggressiveness and immune-escape.
d-dopachrome tautomerase
macrophage migration inhibitory factor
macrophage migration inhibitory factor inhibitors
neuroblastoma
1. Neuroblastoma (NB)
NB represents the most frequent extracranial pediatric tumor, with an incidence of 10.5 cases per million children among 0 and 14 years of age, in North America and Europe [1]. There are no relevant geographic differences in incidence [1]. However, African American and Native American patients are more likely to have worse outcomes, thus showing ethnic disparities [1]. The majority of NB patients are diagnosed before 5 years of age, with a median age at diagnosis of 19 months [2]. In addition, NB is responsible for 12%–15% of cancer-related mortality in children [1].
NB arises from primordial neural precursor cells of the sympathetic nervous system and mainly develops in the adrenal medulla and/or in the paraspinal sympathetic ganglia of the neck, chest, abdomen or pelvis [3]. About half of patients develop distant metastases and the most frequent metastatic sites are bones, bone marrow, and liver [2].
NB is characterized by heterogeneous clinical course and presentation [4]. While some NBs regress spontaneously, others may aggressively spread [4]. Moreover, signs and symptoms are very different, depending on tumor site and biology and on the eventual presence of metastasis or paraneoplastic syndromes [4].
According to the International NB Staging Series (INSS), which relies on surgical observations, NB is classified by risk level, tumor location and dissemination, and MYCN (proto-oncogene protein) amplification [5]. The International NB Risk Group (INRG) Staging System was more recently designed in order to find homogeneous pretreatment risk groups, considering clinical criteria and tumor imaging [6]. The INRG classification takes into account several factors, such as tumor stage and differentiation, patient age, histology, MYCN oncogene status, DNA ploidy, and segmental chromosomal anomalies, in particular chromosome 11q aberration [6]. According to the INRG classification, the patients are stratified in groups with different outcomes and risks including very low, low, intermediate, and high risk [6]. While very low-risk patients have a 5-year event-free survival (EFS) higher than 85%, high-risk patients show a 5-year EFS less than 50% [6].
According to the risk classification, there are different therapeutic approaches for NB patients, such as observation, surgical tumor removal, chemo- and radiotherapy, autologous hematopoietic stem cell transplantation (AHSCT), differentiation therapy, and immunotherapy [7]. In particular, the antidisialoganglioside (anti-GD2) immunotherapy has recently been successfully incorporated into the standard of care treatment for high-risk NB patients [8]. Moreover, a recent randomized clinical trial (NCT00567567) has demonstrated that tandem autologous stem cell transplant with thiotepa/cyclophosphamide followed by carboplatin/etoposide/melphalan resulted in a significantly better EFS than single transplantation with carboplatin/etoposide/melphalan in high-risk NB patients under 30 years of age [9]. Several innovative strategies aimed at targeting the tumor microenvironment, the noradrenaline transporter, and the genetic pathways are being developed with promising effects in NB diagnosis and treatment [7].
2. The Macrophage Migration Inhibitory Factor (MIF) Family of Cytokines
2.1. MIF
Macrophage Migration Inhibitory Factor (MIF) is a multipotent cytokine discovered in 1966 and is characterized as a T cell-derived mediator, with the peculiar property to inhibit the random movement of macrophages [10].
However, MIF is also expressed by different cell lines such as epithelial, endothelial, and immune cells [11]. Unlike many other cytokines that are secreted upon antigenic stimulation, MIF is constantly expressed and stored in intracellular pools [11]. In addition to cytokine function, MIF also exhibits pleiotropic characteristics of enzyme, hormone, and chaperone protein [11]. MIF plays an important role in the regulation of different physiological functions. Harper et al. reported that MIF regulates energy metabolism through its neuroendocrine effects on insulin signaling pathways in the pancreas, muscle, and adipocytes [12]. Furthermore, MIF has been observed to have effects on the hypothalamic–pituitary–adrenal (HPA) axis. In vivo studies in rodents indicate that MIF is released in association with adrenocorticotropin (ACTH) from the pituitary gland during a period of physiological stress [13]. It was reported that MIF-knockout (KO) mice are fertile, their progeny develop and age normally without showing spontaneous diseases [11]. Moreover, Toso et al. reported in a model that MIF knockout (KO) mice or mice treated with anti-MIF show normal blood glucose levels, lactate response, and liver glycogen content after the administration of endotoxin or TNF-α [14].
MIF activates the signaling complex by binding the protein cluster of differentiation (CD) 74 and the signal transducer CD44 or by interacting with the intracellular receptor JAB1 [15]. At the same time, MIF can activate the family of CXC chemokine receptors (CXCR2, CXCR4, and CXCR7) [15]. The role of the interaction between MIF and CXCR7 through Akt-dependent signaling has been recently studied [16]. MIF receptors establish four different receptor complexes to transduce the signaling pathway: CD74/CD44, CD74/CXCR2, CD74/CXCR4, and CD74/CXCR4/CXCR7 [17]. Genetic ablation or anti-CD74 treatment abolishes MIF signaling in CD44-, CXCR2-, CXCR4-, or CXCR7-expressing cells [17]. CD44 is a co-receptor of CD74 and is pivotal for MIF signal transduction [18]. Upon engagement of MIF with the CD74–CD44 complex, the Src kinase is activated [17], leading to the phosphorylation of the extracellular-signal-regulated kinase ½ (ERK1/2) and the inhibition of tumor suppressor protein 53 (p53) expression [19]. The phosphorylation of extracellular signal-regulated kinase-1 (ERK1) and extracellular signal-regulated kinase-2 (ERK2) of mitogen-activated protein (MAP) kinases is closely related to the transduction of the MIF signal and its interaction with the CD74/CD44 complex [19]. The interaction of MIF and CD74 also promotes the activation of the AKT pathway through the mediation of kinases SRC and PI3K [19][20][21].
The activation of AKT leads to the phosphorylation and inactivation of the pro-apoptotic proteins BCL2 associated agonist of cell death (BAD) and Bcl-2-associated X protein (BAX), allowing the cells to resist apoptosis [20]. Furthermore, in lymphoid cells, the activation of AKT, related to an increase of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) function, promotes the expression of the anti-apoptotic proteins Bcl-xL and Bcl-2 [20].
2.2. d-Dopachrome Tautomerase (DDT)
The second member of the MIF family, named DDT or MIF-2, was described in 1997 [22]. Located on the human chromosome 22q11.23, DDT has a homology of 34% with MIF and has a common homotrimer structure [23]. Both homologs have common biological characteristics, such as the enzymatic activity represented by a catalytic proline residue [15]. Like MIF, DDT is able to interact with the CD74 receptor. However, the absence of a binding domain does not allow interaction with CXCR2.
Similarly to MIF, DDT activates the cascade of the MAP kinase ERK1/2 via the activation of the CD74/CD44 complex. This interaction leads to the activation of protein kinase A (PKA), which subsequently phosphorylates SCR and mediates ERK1/2 [17].
It is worth mentioning that DDT was also shown to bind JAB1/CSN5 intracellularly [17].
3. The Role of MIF Family in Cancer
3.1. MIF and Cancer
We and others have shown that MIF and DDT are involved in several diseases of different origins such as immunoinflammatory and autoimmune diseases, neurodegenerative and neuropsychiatric diseases, and cancer [24][25][26][27][28][29][30][31].
In addition, evidence generated during the last 15 years has also supported a pro-oncogenic role of MIF in certain types of cancers [29][30][31]. MIF may upregulate several tumorigenic processes, including tumor growth, invasiveness, and angiogenesis [32] primarily, but not exclusively, through its angiogenetic action, its AKT mediated antiapoptotic effects, and the inhibition of p53 function.
Strictly related to vessel growth and cell proliferation is the ability of MIF to activate hypoxia-induced factor-1α (HIF-1α) [33] through the extracellular and intracellular environment [33]. When MIF binds to CD74, it leads to the direct HIF1α activation, while in the intracellular domain, MIF binds Jab1/CSN5, which regulates the functionality of HIF1α by counteracting its hydroxylation and leads to the expression of pro-angiogenic factors, such as IL-8 and vascular endothelial growth factor (VEGF) [33]. At the intracellular level, MIF is also able to modulate AP-1 activity and the cell cycle, synergistically with Jab1/CSN5 interaction, inactivating the tumor suppressor p53 [34] (Figure 1).
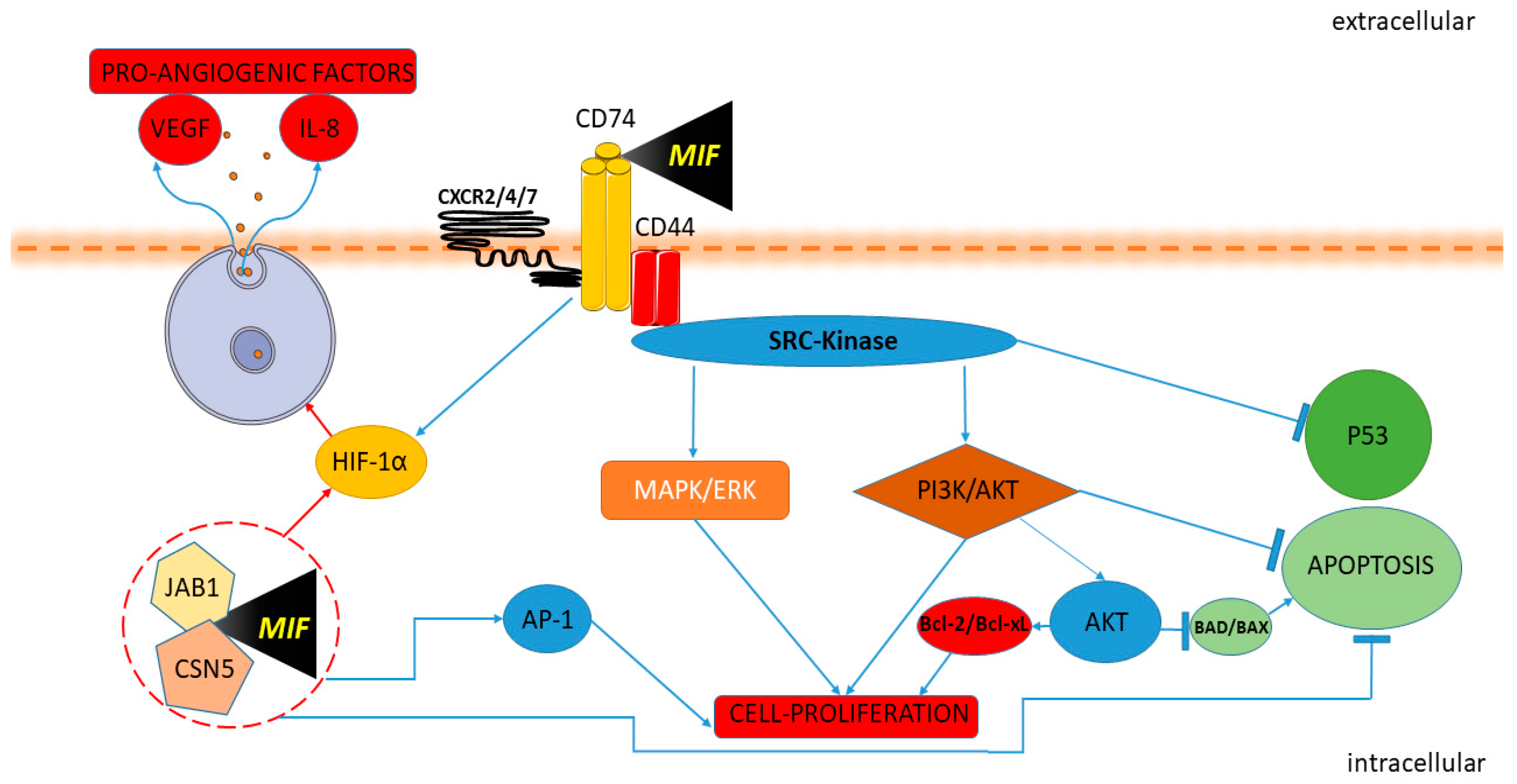
Figure 1. Macrophage migration inhibitory factor (MIF) and cancer pathway. MIF activates the signaling complex binding CD74, CD44 and the chemokine receptors CXCR2, CXCR4, and CXCR7. The Protein Tyrosine Kinase Src is activated by MIF. Upon the engagement of the CD74-CD44 complex, the Src kinase is activated, leading to phosphorylation of the extracellular-signal-regulated kinase ERK/MAP and inhibition of p53 expression. The interaction of MIF and CD74 also promotes the activation of the AKT/PI3K pathway with the consequent inactivation of the pro-apoptotic proteins BAD and BAX and promotes the expression of the anti-apoptotic proteins Bcl-xL and Bcl-2. MIF induces HIF1α activation through extracellular and intracellular interaction. MIF binding to CD74 leads to HIF1α activation. In the intracellular domain, MIF binds Jab1/CSN5, activating HIF1α and leading to the expression of proangiogenic factors IL-8 and VEGF. MIF modulates AP-1 activity and cell proliferation with Jab1/CSN5 interaction and inactivating p53.
Though endowed with proinflammatory activities, several lines of evidence also suggest that at the tumor site MIF may act as a soluble immune checkpoint inhibitor, thus favoring the establishment of immune-evasion in the microenvironment [15] via induction of myeloid-derived suppressor cells [35] and inhibition of T cells activation [36], M1 polarization [37] and reduction of natural killer (NK) cell cytotoxicity [38].
MIF may be implicated in certain forms of tumorigenesis as suggested by genetic studies showing that the single nucleotide polymorphism (SNP) -173 G/C (rs755622) on MIF gene is associated to MIF hyperproduction and correlates with cancer [39].
A meta-analysis by Vera et al. has shown an association between the -173C MIF promoter polymorphism and an increased risk of cancer, particularly for prostate cancer and other solid tumors [40]. Moreover, the genetic polymorphism MIF-173 was associated with a higher risk of early cervical cancer and lymph node metastasis [41] and also with the risk of gastrointestinal cancer and hematological malignancy [42]. In addition, Lin et al. found that MIF rs755622 polymorphism correlated with breast cancer susceptibility in Chinese population, particularly in elderly patients [43].
Numerous preclinical and clinical studies have demonstrated that MIF is overexpressed and may correlate with tumor aggressiveness in many different types of human cancers such as prostate , bladder, and kidney cancer [30], cervical cancer [20], ovarian cancer [44][45], breast cancer [29], gastric cancer [29], hepatocellular carcinoma [46], colon cancer [47][48], pancreatic cancer [49][50], gallbladder cancer [51], lung cancer [29][52], melanoma [31], head and neck cancer [29], acute myeloid leukemia [53][54], glioblastoma [15][55][56], and NB [57][58][59][60]. Moreover, elevated MIF expression is correlated with a worse patient overall survival in a large variety of cancers such as breast cancer [29], gastric cancer [29], hepatocellular carcinoma [61], pancreatic cancer [49][50], metastatic melanoma [31], head and neck cancer [29], esophageal squamous cell carcinoma [62], acute myeloid leukemia [53], glioblastoma [29][56], and NB [57]. However, conflicting results have also been reported and other studies have shown that, in other types of tumors, endogenous MIF may possess a beneficial anticancer activity.
For example, low nuclear MIF expression conferred a poor prognosis to patients with lung adenocarcinoma [52]. Differently, high levels of MIF were correlated with reduced drug responsiveness and with poorer outcomes in lung cancer patients [29]. Conflicting results on the role of MIF in breast cancers have been reported with a study claiming to a correlation between positive MIF expression and a better overall and recurrence-free survival [63] and others demonstrating that positive MIF expression levels correlated with a worse prognosis [64][65]. Contradictory results also exist for colon cancer patients [47] with one study showing that MIF expression was associated with tumor grade and hepatic metastases [66] and another indicating that the elevated MIF expression correlated with better survival in Dukes C or D colorectal tumors [67].
It has also been shown that increased MIF expression in tumor-infiltrating lymphocytes (TILs) within tumor microenvironments was associated with better outcomes in nasopharyngeal carcinoma (NPC) patients [68].
Taken as a whole, these data seem to indicate that the MIF possesses both pro and antioncogenic activities that may depend on the phenotype and site of the tumor and possibly the genetic background of the patients and other yet unidentified cofactors. The data seem to indicate that, for certain types of tumors, both local expression of MIF and its circulating blood levels seem to be promising prognostic and predictive biomarkers and tailored anti-MIF therapies may be beneficial [15].
3.2. DDT (MIF2) and Cancer
There are only a few studies on the role of DDT in cancer. It has been shown that the knockdown of DDT and MIF in the pancreatic cell line, PANC-1, correlated with reduced activation of ERK1/2 and AKT, augmented p53 expression, and inhibited tumor growth in vitro and in vivo [15][69]. The DDT interaction with CD74 stimulates the expression of VEGF and CXCL8 and counteracts the 5’ AMP-activated protein kinase (AMPK) activation in human non-small cell lung carcinoma [70][71]. Interestingly, in contrast to the ability of MIF and DDT to activate AMPK in non-transformed cells, they cooperatively inhibited the activation of AMPK in LKB1 mutant human non-small cell lung cancer (NSCLC) cell lines [71]. Furthermore, treatment with the dual inhibitor of MIF and DDT, 4-iodo-6-phenylpyrimidine (4-IPP), decreased in vitro proliferation and in vivo tumor growth in a mouse xenograft model [69]. Moreover, in the melanoma cancer cell line B16F10, treatment with small interfering RNAs (siRNA)/DDT suppressed cell proliferation and stimulated apoptosis, and in a xenograft model treatment with anti-DDT antibodies reduced tumor growth [72]. DDT has also been reported in colorectal cancer, showing to regulate the transcriptional factor β-catenin, in a manner partly dependent on COX-2 expression [73]. In DDT-deficient colorectal cells, the β-catenin expression is reduced [73]. These data suggest that DDT may play an overlapping role with MIF, thus suggesting possible therapeutic actions aimed at inhibiting the two homologs.
References
- Irwin, M.S.; Park, J.R. Neuroblastoma: Paradigm for precision medicine. Pediatr. Clin. North Am. 2015, 62, 225–256.
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386.
- Aygun, N. Biological and Genetic Features of Neuroblastoma and Their Clinical Importance. Curr. Pediatr. Rev. 2018, 14, 73–90.
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120.
- Salazar, B.M.; Balczewski, E.A.; Ung, C.Y.; Zhu, S. Neuroblastoma, a paradigm for big data science in pediatric oncology. Int. J. Mol. Sci. 2017, 18, 37.
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297.
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078.
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334.
- Park, J.R.; Kreissman, S.G.; London, W.B.; Naranjo, A.; Cohn, S.L.; Hogarty, M.D.; Tenney, S.C.; Haas-Kogan, D.; Shaw, P.J.; Kraveka, J.M.; et al. Effect of tandem autologous stem cell transplant vs single transplant on event-free survival in patients with high-risk neuroblastoma: A randomized clinical trial. JAMA - J. Am. Med. Assoc. 2019, 322, 746–755.
- Bloom, B.R.; Bennett, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 1966, 153, 80–82.
- Stosic-Grujicic, S.; Stojanovic, I.; Nicoletti, F. MIF in autoimmunity and novel therapeutic approaches. Autoimmun. Rev. 2009, 8, 244–249.
- Harper, J.M.; Wilkinson, J.E.; Miller, R.A. Macrophage migration inhibitory factor-knockout mice are long lived and respond to caloric restriction. FASEB J. 2010, 24, 2436–2442.
- Bucala, R. MIF rediscovered: Cytokine, pituitary hormone, and glucocorticoid-induced regulator of the immune response. FASEB J. 1996, 10, 1607–1613.
- Toso, C.; Emamaullee, J.A.; Merani, S.; Shapiro, A.M.J. The role of macrophage migration inhibitory factor on glucose metabolism and diabetes. Diabetologia 2008, 51, 1937–1946.
- Mangano, K.; Mazzon, E.; Basile, M.S.; Di Marco, R.; Bramanti, P.; Mammana, S.; Petralia, M.C.; Fagone, P.; Nicoletti, F. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget 2018, 9, 17951–17970.
- Chatterjee, M.; Borst, O.; Walker, B.; Fotinos, A.; Vogel, S.; Seizer, P.; Mack, A.; Alampour-Rajabi, S.; Rath, D.; Geisler, T.; et al. Macrophage migration inhibitory factor limits activation-induced apoptosis of platelets via CXCR7-dependent Akt signaling. Circ. Res. 2014, 115, 939–949.
- Jankauskas, S.S.; Wong, D.W.L.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving complexity of MIF signaling. Cell. Signal. 2019, 57, 76–88.
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 Is the Signaling Component of the Macrophage Migration Inhibitory Factor-CD74 Receptor Complex. Immunity 2006, 25, 595–606.
- Lue, H.; Kapurniotu, A.; Fingerle-Rowson, G.; Roger, T.; Leng, L.; Thiele, M.; Calandra, T.; Bucala, R.; Bernhagen, J. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell. Signal. 2006, 18, 688–703.
- Nobre, C.C.G.; de Araújo, J.M.G.; de Fernandes, T.A.A.M.; Cobucci, R.N.O.; Lanza, D.C.F.; Andrade, V.S.; Fernandes, J.V. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathol. Oncol. Res. 2017, 23, 235–244.
- Mammana, S.; Bramanti, P.; Mazzon, E.; Cavalli, E.; Basile, M.S.; Fagone, P.; Petralia, M.C.; McCubrey, J.A.; Nicoletti, F.; Mangano, K. Preclinical evaluation of the PI3K/Akt/mTOR pathway in animal models of multiple sclerosis. Oncotarget 2018, 9, 8263–8277.
- Sugimoto, H.; Taniguchi, M.; Nakagawa, A.; Tanaka, I.; Suzuki, M.; Nishihira, J. Crystallization and preliminary X-ray analysis of human D-dopachrome tautomerase. J. Struct. Biol. 1997, 120, 105–108.
- Kang, I.; Bucala, R. The immunobiology of MIF: Function, genetics and prospects for precision medicine. Nat. Rev. Rheumatol. 2019, 15, 427–437.
- Petralia, M.C.; Battaglia, G.; Bruno, V.; Pennisi, M.; Mangano, K.; Lombardo, S.D.; Fagone, P.; Cavalli, E.; Saraceno, A.; Nicoletti, F.; et al. The Role of Macrophage Migration Inhibitory Factor in Alzheimer′s Disease: Conventionally Pathogenetic or Unconventionally Protective? Molecules 2020, 25, 291.
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Basile, M.S.; Lenzo, V.; Quattropani, M.C.; Bendtzen, K.; Nicoletti, F. Pathogenic contribution of the Macrophage migration inhibitory factor family to major depressive disorder and emerging tailored therapeutic approaches. J. Affect. Disord. 2020, 263, 15–24.
- Lombardo, S.D.; Mazzon, E.; Mangano, K.; Basile, M.S.; Cavalli, E.; Mammana, S.; Fagone, P.; Nicoletti, F.; Petralia, M.C. Transcriptomic analysis reveals involvement of the macrophage migration inhibitory factor gene network in duchenne muscular dystrophy. Genes 2019, 10, 939.
- Cavalli, E.; Mazzon, E.; Basile, M.S.; Mangano, K.; Di Marco, R.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Upregulated Expression of Macrophage Migration Inhibitory Factor, Its Analogue D-Dopachrome Tautomerase, and the CD44 Receptor in Peripheral CD4 T Cells from Clinically Isolated Syndrome Patients with Rapid Conversion to Clinical Defined Multiple Sclerosi. Medicina (B. Aires) 2019, 55, 667.
- Fagone, P.; Mazzon, E.; Cavalli, E.; Bramanti, A.; Petralia, M.C.M.C.M.C.M.C.; Mangano, K.; Al-Abed, Y.; Bramati, P.; Nicoletti, F. Contribution of the macrophage migration inhibitory factor superfamily of cytokines in the pathogenesis of preclinical and human multiple sclerosis: In silico and in vivo evidences. J. Neuroimmunol. 2018, 322, 46–56.
- Guda, M.R.; Rashid, M.A.; Asuthkar, S.; Jalasutram, A.; Caniglia, J.L.; Tsung, A.J.; Velpula, K.K. Pleiotropic role of macrophage migration inhibitory factor in cancer. Am. J. Cancer Res. 2019, 9, 2760–2773.
- Penticuff, J.C.; Woolbright, B.L.; Sielecki, T.M.; Weir, S.J.; Taylor, J.A. MIF family proteins in genitourinary cancer: Tumorigenic roles and therapeutic potential. Nat. Rev. Urol. 2019, 16, 318–328.
- Soumoy, L.; Kindt, N.; Ghanem, G.; Saussez, S.; Journe, F. Role of Macrophage Migration Inhibitory Factor (MIF) in Melanoma. Cancers 2019, 11, 529.
- Kindt, N.; Journe, F.; Laurent, G.; Saussez, S. Involvement of macrophage migration inhibitory factor in cancer and novel therapeutic targets (Review). Oncol. Lett. 2016, 12, 2247–2253.
- Shackleford, T.J.; Claret, F.X. JAB1/CSN5: A new player in cell cycle control and cancer. Cell Div. 2010, 5, 26.
- Kleemann, R.; Hausser, A.; Geiger, G.; Mischke, R.; Burger-Kentischer, A.; Flieger, O.; Johannes, F.J.; Roger, T.; Calandra, T.; Kapurniotu, A.; et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 2000, 408, 211–216.
- Simpson, K.D.; Templeton, D.J.; Cross, J.V. Macrophage Migration Inhibitory Factor Promotes Tumor Growth and Metastasis by Inducing Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. J. Immunol. 2012, 189, 5533–5540.
- Yan, X.; Orentas, R.J.; Johnson, B.D. Tumor-derived macrophage migration inhibitory factor (MIF) inhibits T lymphocyte activation. Cytokine 2006, 33, 188–198.
- Ghoochani, A.; Schwarz, M.A.; Yakubov, E.; Engelhorn, T.; Doerfler, A.; Buchfelder, M.; Bucala, R.; Savaskan, N.E.; Eyüpoglu, I.Y. MIF-CD74 signaling impedes microglial M1 polarization and facilitates brain tumorigenesis. Oncogene 2016, 35, 6246–6261.
- Apte, R.S.; Sinha, D.; Mayhew, E.; Wistow, G.J.; Niederkorn, J.Y. Cutting edge: Role of macrophage migration inhibitory factor in inhibiting NK cell activity and preserving immune privilege. J. Immunol. 1998, 160, 5693–5696.
- Illescas, O.; Gomez-Verjan, J.C.; García-Velázquez, L.; Govezensky, T.; Rodriguez-Sosa, M. Macrophage Migration Inhibitory Factor -173 G/C Polymorphism: A Global Meta-Analysis across the Disease Spectrum. Front. Genet. 2018, 9, 55.
- Vera, P.L.; Meyer-Siegler, K.L. Association between macrophage migration inhibitory factor promoter region polymorphism (-173 G/C) and cancer: A meta-analysis. BMC Res. Notes 2011, 4, 395.
- Wu, S.; Lian, J.; Tao, H.; Shang, H.; Zhang, L. Correlation of macrophage migration inhibitory factor gene polymorphism with the risk of early-stage cervical cancer and lymphatic metastasis. Oncol. Lett. 2011, 2, 1261–1267.
- Tong, X.; Zheng, B.; Tong, Q.; Liu, S.; Peng, S.; Yang, X.; Fan, H. The MIF -173G/C gene polymorphism increase gastrointestinal cancer and hematological malignancy risk: Evidence from a meta-analysis and FPRP test. Int. J. Clin. Exp. Med. 2015, 8, 15949–15957.
- Lin, S.; Wang, M.; Liu, X.; Zhu, W.; Guo, Y.; Dai, Z.Z.; Yang, P.; Tian, T.; Dai, C.; Zheng, Y.; et al. Association of genetic polymorphisms in MIF with breast cancer risk in Chinese women. Clin. Exp. Med. 2017, 17, 395–401.
- Krockenberger, M.; Dombrowski, Y.; Weidler, C.; Ossadnik, M.; Hönig, A.; Häusler, S.; Voigt, H.; Becker, J.C.; Leng, L.; Steinle, A.; et al. Macrophage Migration Inhibitory Factor Contributes to the Immune Escape of Ovarian Cancer by Down-Regulating NKG2D. J. Immunol. 2008, 180, 7338–7348.
- Hagemann, T.; Robinson, S.C.; Thompson, R.G.; Charles, K.; Kulbe, H.; Balkwill, F.R. Ovarian cancer cell-derived migration inhibitory factor enhances tumor growth, progression, and angiogenesis. Mol. Cancer Ther. 2007, 6, 1993–2002.
- Huang, X.H.; Jian, W.H.; Wu, Z.F.; Zhao, J.; Wang, H.; Li, W.; Xia, J.T. Small interfering RNA (siRNA)-mediated knockdown of macrophage migration inhibitory factor (MIF) suppressed cyclin D1 expression and hepatocellular carcinoma cell proliferation. Oncotarget 2014, 5, 5570–5580.
- Gordon-Weeks, A.N.; Lim, S.Y.; Yuzhalin, A.E.; Jones, K.; Muschel, R. Macrophage migration inhibitory factor: A key cytokine and therapeutic target in colon cancer. Cytokine Growth Factor Rev. 2015, 26, 451–461.
- Mamoori, A.; Wahab, R.; Vider, J.; Gopalan, V.; Lam, A.K. The tumour suppressor effects and regulation of cancer stem cells by macrophage migration inhibitory factor targeted miR-451 in colon cancer. Gene 2019, 697, 165–174.
- Wang, D.; Wang, R.; Huang, A.; Fang, Z.; Wang, K.; He, M.; Xia, J.T.; Li, W. Upregulation of macrophage migration inhibitory factor promotes tumor metastasis and correlates with poor prognosis of pancreatic ductal adenocarcinoma. Oncol. Rep. 2018, 40, 2628–2636.
- Funamizu, N.; Hu, C.; Lacy, C.; Schetter, A.; Zhang, G.; He, P.; Gaedcke, J.; Ghadimi, M.B.; Ried, T.; Yfantis, H.G.; et al. Macrophage migration inhibitory factor induces epithelial to mesenchymal transition, enhances tumor aggressiveness and predicts clinical outcome in resected pancreatic ductal adenocarcinoma. Int. J. Cancer 2013, 132, 785–794.
- Subbannayya, T.; Leal-Rojas, P.; Barbhuiya, M.A.; Raja, R.; Renuse, S.; Sathe, G.; Pinto, S.M.; Syed, N.; Nanjappa, V.; Patil, A.H.; et al. Macrophage migration inhibitory factor - a therapeutic target in gallbladder cancer. BMC Cancer 2015, 15, 843.
- Kamimura, A.; Kamachi, M.; Nishihira, J.; Ogura, S.; Isobe, H.; Dosaka-Akita, H.; Ogata, A.; Shindoh, M.; Ohbuchi, T.; Kawakami, Y. Intracellular distribution of macrophage migration inhibitory factor predicts the prognosis of patients with adenocarcinoma of the lung. Cancer 2000, 89, 334–341.
- Falantes, J.F.; Trujillo, P.; Piruat, J.I.; Calderón, C.; Márquez-Malaver, F.J.; Martín-Antonio, B.; Millán, A.; Gómez, M.; González, J.; Martino, M.L.; et al. Overexpression of GYS1, MIF, and MYC Is associated with adverse outcome and poor response to azacitidine in myelodysplastic syndromes and acute myeloid Leukemia. Clin. Lymphoma, Myeloma Leuk. 2015, 15, 236–244.
- Abdul-Aziz, A.M.; Shafat, M.S.; Sun, Y.; Marlein, C.R.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Zhou, Z.; Collins, A.; Bowles, K.M.; et al. HIF1α drives chemokine factor pro-tumoral signaling pathways in acute myeloid leukemia. Oncogene 2018, 37, 2676–2686.
- Presti, M.; Mazzon, E.; Basile, M.S.; Petralia, M.C.; Bramanti, A.; Colletti, G.; Bramanti, P.; Nicoletti, F.; Fagone, P. Overexpression of macrophage migration inhibitory factor and functionally-related genes, D-DT, CD74, CD44, CXCR2 and CXCR4, in glioblastoma. Oncol. Lett. 2018, 16, 2881–2886.
- Ha, W.; Sevim-Nalkiran, H.; Zaman, A.M.; Matsuda, K.; Khasraw, M.; Nowak, A.K.; Chung, L.; Baxter, R.C.; McDonald, K.L. Ibudilast sensitizes glioblastoma to temozolomide by targeting Macrophage Migration Inhibitory Factor (MIF). Sci. Rep. 2019, 9, 1–10.
- Cavalli, E.; Mazzon, E.; Mammana, S.; Basile, M.S.; Lombardo, S.D.; Mangano, K.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Overexpression of macrophage migration inhibitory factor and its homologue d-dopachrome tautomerase as negative prognostic factor in neuroblastoma. Brain Sci. 2019, 9, 284.
- Ren, Y.; Chan, H.M.; Li, Z.; Lin, C.; Nicholls, J.; Chen, C.F.; Lee, P.Y.; Lui, V.; Bacher, M.; Tam, P.K.H. Upregulation of macrophage migration inhibitory factor contributes to induced N-Myc expression by the activation of ERK signaling pathway and increased expression of interleukin-8 and VEGF in neuroblastoma. Oncogene 2004, 23, 4146–4154.
- Zhou, Q.; Yan, X.; Gershan, J.; Orentas, R.J.; Johnson, B.D. Expression of Macrophage Migration Inhibitory Factor by Neuroblastoma Leads to the Inhibition of Antitumor T Cell Reactivity In Vivo. J. Immunol. 2008, 181, 1877–1886.
- Bin, Q.; Johnson, B.D.; Schauer, D.W.; Casper, J.T.; Orentas, R.J. Production of macrophage migration inhibitory factor by human and murine neuroblastoma. Tumour Biol. 2002, 23, 123–129.
- Zhao, Y.-M.; Wang, L.; Dai, Z.; Wang, D.-D.; Hei, Z.-Y.; Zhang, N.; Fu, X.-T.; Wang, X.-L.; Zhang, S.-C.; Qin, L.-X.; et al. Validity of plasma macrophage migration inhibitory factor for diagnosis and prognosis of hepatocellular carcinoma. Int. J. Cancer 2011, 129, 2463–2472.
- Zhang, L.; Ye, S.B.; Ma, G.; Tang, X.F.; Chen, S.P.; He, J.; Liu, W.L.; Xie, D.; Zeng, Y.X.; Li, J. The expressions of MIF and CXCR4 protein in tumor microenvironment are adverse prognostic factors in patients with esophageal squamous cell carcinoma. J. Transl. Med. 2013, 11, 60.
- Verjans, E.; Noetzel, E.; Bektas, N.; Schütz, A.K.; Lue, H.; Lennartz, B.; Hartmann, A.; Dahl, E.; Bernhagen, J. Dual role of macrophage migration inhibitory factor (MIF) in human breast cancer. BMC Cancer 2009, 9, 230.
- Wang, P.; Liu, J.; Song, Y.; Liu, Q.; Wang, C.; Qian, C.; Zhang, S.; Zhu, W.; Yang, X.; Wan, F.; et al. Screening of immunosuppressive factors for biomarkers of breast cancer malignancy phenotypes and subtype-specific targeted therapy. PeerJ 2019, 7, e7197.
- Xu, X.; Wang, B.; Ye, C.; Yao, C.; Lin, Y.; Huang, X.; Zhang, Y.; Wang, S. Overexpression of macrophage migration inhibitory factor induces angiogenesis in human breast cancer. Cancer Lett. 2008, 261, 147–157.
- He, X.-X.; Chen, K.; Yang, J.; Li, X.-Y.; Gan, H.-Y.; Liu, C.-Y.; Coleman, T.R.; Al-Abed, Y. Macrophage migration inhibitory factor promotes colorectal cancer. Mol. Med. 2009, 15, 1–10.
- Legendre, H.; Decaestecker, C.; Nagy, N.; Hendlisz, A.; Schüring, M.P.; Salmon, I.; Gabius, H.J.; Pector, J.C.; Kiss, R. Prognostic values of galectin-3 and the macrophage migration inhibitory factor (MIF) in human colorectal cancers. Mod. Pathol. 2003, 16, 491–504.
- Li, J.; Mo, H.Y.; Xiong, G.; Zhang, L.; He, J.; Huang, Z.F.; Liu, Z.W.; Chen, Q.Y.; Du, Z.M.; Zheng, L.M.; et al. Tumor microenvironment macrophage inhibitory factor directs the accumulation of interleukin-17-producing tumor-infiltrating lymphocytes and predicts favorable survival in nasopharyngeal carcinoma patients. J. Biol. Chem. 2012, 287, 35484–35495.
- Guo, D.; Guo, J.; Yao, J.; Jiang, K.; Hu, J.; Wang, B.; Liu, H.; Lin, L.; Sun, W.; Jiang, X. D-dopachrome tautomerase is over-expressed in pancreatic ductal adenocarcinoma and acts cooperatively with macrophage migration inhibitory factor to promote cancer growth. Int. J. Cancer 2016, 139, 2056–2067.
- Coleman, A.M.; Rendon, B.E.; Zhao, M.; Qian, M.-W.; Bucala, R.; Xin, D.; Mitchell, R.A. Cooperative Regulation of Non-Small Cell Lung Carcinoma Angiogenic Potential by Macrophage Migration Inhibitory Factor and Its Homolog, d-Dopachrome Tautomerase. J. Immunol. 2008, 181, 2330–2337.
- Brock, S.E.; Rendon, B.E.; Yaddanapudi, K.; Mitchell, R.A. Negative regulation of AMP-activated protein kinase (AMPK) activity by macrophage migration inhibitory factor (MIF) family members in non-small cell lung carcinomas. J. Biol. Chem. 2012, 287, 37917–37925.
- Kobold, S.; Merk, M.; Hofer, L.; Peters, P.; Bucala, R.; Endres, S. The macrophage migration inhibitory factor (MIF)-homologue D-dopachrome tautomerase is a therapeutic target in a murine melanoma model. Oncotarget 2014, 5, 103–107.
- Xin, D.; Rendon, B.E.; Zhao, M.; Winner, M.; Coleman, A.M.G.; Mitchell, R.A. The MIF homologue d-dopachrome tautomerase promotes COX-2 expression through β-catenin-dependent and -independent mechanisms. Mol. Cancer Res. 2010, 8, 1601–1609.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
835
Revisions:
2 times
(View History)
Update Date:
19 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No