 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kudakwashe Mhandire | + 3028 word(s) | 3028 | 2021-11-07 06:07:33 | | | |
| 2 | Lindsay Dong | + 279 word(s) | 3307 | 2021-11-18 10:37:15 | | |
Video Upload Options
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a curative option in the treatment of aggressive malignant and non-malignant blood disorders. However, the benefits of allo-HSCT can be compromised by graft-versus-host disease (GvHD), a prevalent and morbid complication of allo-HSCT. GvHD occurs when donor immune cells mount an alloreactive response against host antigens due to histocompatibility differences between the donor and host, which may result in extensive tissue injury. The reprogramming of cellular metabolism is a feature of GvHD that is associated with the differentiation of donor CD4+ cells into the pathogenic Th1 and Th17 subsets along with the dysfunction of the immune-suppressive protective T regulatory cells (Tregs). The activation of glycolysis and glutaminolysis with concomitant changes in fatty acid oxidation metabolism fuel the anabolic activities of the proliferative alloreactive microenvironment characteristic of GvHD. Thus, metabolic therapies such as glycolytic enzyme inhibitors and fatty acid metabolism modulators are a promising therapeutic strategy for GvHD.
1. Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) has the potential to cure multiple hematological disorders, including aggressive malignancies [1]. The curative potential of allo-HSCT, the graft-versus-leukemia (GvL) effect, can be mediated by several immune subsets [2][3][4]. However, these cell populations may also trigger graft-versus-host disease (GvHD), a morbid and prevalent barrier to allo-HSCT [5][6]. CD4+ T cells are at the center of GvHD pathogenesis. Activated CD4+ T cells differentiate into a plethora of cytokine-secreting T helper (Th) cells including Th1, Th2, Th9, Th17, and regulatory T cells (Tregs) [7][8][9]. Pro-inflammatory Th1 and Th17 CD4+ cells direct GvHD targeting the skin, lungs, gastrointestinal tract, and other tissues [4][10][11][12]. By contrast, regulatory CD4+ Forkhead box protein P3 (Foxp3) + cells (Tregs) are capable of ameliorating GvHD through immunosuppressive mechanisms [13][14][15][16][17]. Alloreactivity induces a metabolic shift in immune cells from a quiescent oxidative phosphorylation (OXPHOS) dominant state to an anabolic profile featuring aerobic glycolysis as the main energy source. In addition, glutamine and fatty acid metabolism provide intermediates for the tri-carboxylic acid (TCA) cycle which fuels the OXPHOS activity seen in early T cell activation [10][18][19][20]. The increased glycolytic flux supports the proliferation of the pathogenic Th1 and Th17 cells. The resultant high T effector/Treg ratio facilitates GvHD pathogenesis in target tissues [9][10][13][21].
There are several forms of GvHD, including acute, chronic, late acute, and overlap syndrome, and some of the biological and pathophysiological features are shared by these entities [4][5]. Acute GvHD exhibits systemic inflammation and tissue destruction in the skin, liver, and gastrointestinal tract that involves alloreactive donor T-cell-mediated cytotoxicity [4][11]. The pathophysiology of cGvHD is complex and thought to involve the interplay between dysregulated innate and adaptive immune cell populations, as well as tissue fibrosis and organ damage [6][22]. Acute GvHD is the main risk factor of chronic GvHD and together they negatively impact on the success of allo-HSCT [23][24][25][26].
Standard GvHD prophylactic measures involve peri-transplant immunosuppression directed primarily at the T cells compartment, including anti-thymocyte globulin (ATG), alemtuzumab, alpha-beta depletion, post-transplant cyclophosphamide (PTCy), calcineurin inhibitors, and methotrexate [27][28][29][30][31]. Despite prophylaxis, the prevalence of acute and chronic GvHD is high. First-line therapeutic interventions aim to provide global immune suppression via the administration of systemic corticosteroids with or without calcineurin inhibitors [27][32]. Unfortunately, these measures increase the risk of infection, relapse, and secondary malignancies [32][33]. In addition, systemic corticosteroids have many side effects and are ineffective for many patients with acute and chronic GvHD, with clinical responses to second-line agents being also being limited [34][35]. An ideal GvHD therapeutic intervention would suppress pathogenic allogeneic immune reactivity while preserving the graft-versus-leukemia (GvL) effect and maintain functional anti-infection immunity [2][36]. Given that malignant cells share cellular metabolism features with alloreactive T cells, metabolic reprogramming represents a promising strategy for the therapeutic targeting of GvHD [7][8][37].
2. T Cell Metabolism in Allo-HSCT
Naïve T cells primarily depend on oxidative phosphorylation. Following allo-HSCT, donor T cells are stimulated by mismatched recipient cell antigens to undergo pro-glycolytic metabolic reprogramming and form allogeneic T effector cells (Teffs) [38][39][40]. During this phase, pyruvate is directed towards lactate synthesis under the influence of pyruvate dehydrogenase kinase 1 (PDHK1) [18][41]. The upregulation of glycolysis supports cell proliferation as glycolytic intermediates feed anabolic pathways, including the pentose phosphate pathway (PPP) [9][42][43]. T cell receptor (TCR) antigen-presenting cell (APC) interaction stimulates the phosphoinositide 3-kinases (PI3K)-protein kinase B-mechanistic target of rapamycin (mTOR) pathway to stabilize the glycolytic microenvironment. Furthermore, protein kinase B (a.k.a. Akt) activates glycolytic enzymes such as hexokinase (HK) and phosphofructokinase (PFK) through phosphorylation [44][45]. Akt also upregulates the expression of the transmembrane glucose transporters (Glut)1 and Glut3 in alloreactive T cells [46][47].
Concomitantly, pyruvate is decarboxylated into acetyl-CoA which is relayed into the TCA cycle to facilitate NADH and flavin adenine dinucleotide (FADH2) synthesis. In the presence of oxygen, NADH and FADH2 fuel ATP generation by shuttling electrons along the electron transport chain in OXPHOS [43][48]. Studies show that both glycolysis and OXPHOS are upregulated in alloreactive cells [20][49]. However, the increase in OXPHOS activity is transient and may characterize early events in GvHD pathogenesis and T cell activation [49][50], perhaps preceding the development of tissue hypoxia. The alloreactive metabolism and cell proliferation form a hypoxic environment that is characterized by the elevated expression of hypoxia-induced factor 1 alpha (HIF-1α) [51][52]. HIF-1α is a transcription factor, which modulates the expression of glycolytic enzymes, such as HK1, and glucose transporters, namely Glut1, and Glut3 [19][51]. The messenger ribonucleic acid (mRNA) expression of other glycolytic enzymes and transporters, such as glyceraldehyde 3-phosphate dehydrogenase (Gapdh), lactate dehydrogenase (Ldh)-a, monocarboxylate transporter (Mtc4) and phosphoglycerate kinase (Pgk1) have also been found to be elevated in alloreactive T cells in GvHD [19][53].
3. T Cell Metabolic Targets for GvHD
3.1. Glycolysis Targets
The metabolic reprogramming from OXPHOS towards glycolysis sustains the proliferation and secretion of pro-inflammatory cytokines in alloreactive T cells [6][8][43]. Our group confirmed through transcriptomic, protein, and metabolic analyses that the CD4 T effector memory (Tem) cells, a pathogenic subset that mediates GvHD in target tissues, is highly glycolytic [53], providing a further rationale for testing glycolytic inhibitors in GvHD. Pharmacological inhibitors of glycolytic enzymes have been previously tested in an aGvHD pre-clinical model [19] and identified glycolysis as an important metabolic target for re-programming alloreactive T cells in aGvHD (Figure 1).
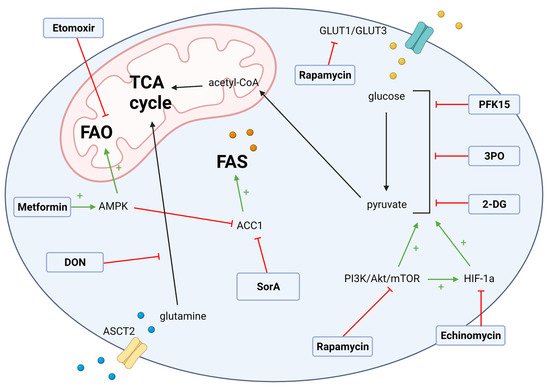
3.2. Fatty Acid Metabolism Targets in GvHD
3.3. Glutamate Metabolism Targets
4. Antigen-Presenting Cell Metabolism in GvHD
4.1. Dendritic Cell Targets
4.2. Macrophage Targets
Macrophages (MCs) are phagocytic antigen-presenting cells with dynamic profiles that reciprocally differentiate into pro-inflammatory M1 and anti-inflammatory M2 subsets [98]. The M1 macrophages are induced by the detection of foreign antigens and pro-inflammatory cytokines (TNF-α, INF-γ), leading to cytokine secretion and the activation of Th1 cells, and the release of reactive nitrogen and oxygen species [99]. By contrast, M2 cells are induced by inflammatory cytokines (IL-4 and IL-13) and Th2 cells to mostly induce immune regulation and the resolution of inflammation [100]. M1 and M2 MCs feature distinct metabolic profiles [98]. The M1 profile is dependent on glycolysis for energy generation, and enforces two breaks in the TCA cycle to produce citrate and succinate [101]. The accumulated citrate generates itaconate, an important antimicrobial agent [102]. Succinate stabilizes HIF-1α, which sustains the glycolytic metabolism of M1 cells [68][103]. Glycolysis feeds the PPP to generate NADPH, which acts as a co-factor of iNOS in NO production [104]. Acetyl-CoA generated by glycolysis is used in FAS. Enhanced FAS is an important feature of M1 metabolism. HIF-1α also suppresses OXPHOS in M1 macrophages, while M2 macrophages rely on OXPHOS for ATP generation. Additionally, in contrast to M1, M2 MCs rely on the TCA cycle [105]. In M2 MCs, FAO and glutamine metabolism are upregulated to provide intermediates for the TCA cycle. Glycolysis and, subsequently, the PPP, are downregulated in M2 macrophages [99].
4.3. Myeloid-Derived Suppressor Cell Targets
4.4. B Cell Ttargets
References
- Phelan R, A.M.; Chen, M. Current Use and Outcome of Hematopoietic Stem Cell Transplantation. 2021. Available online: https://www.cibmtr.org/ReferenceCenter/SlidesReports/SummarySlides/pages/index.aspx (accessed on 22 October 2021).
- Dickinson, A.M.; Norden, J.; Li, S.; Hromadnikova, I.; Schmid, C.; Schmetzer, H.; Jochem-Kolb, H. Graft-versus-Leukemia Effect Following Hematopoietic Stem Cell Transplantation for Leukemia. Front. Immunol. 2017, 8, 496.
- Kolb, H.-J. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood 2008, 112, 4371–4383.
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561.
- Blazar, B.R.; Murphy, W.J.; Abedi, M. Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 2012, 12, 443–458.
- Zeiser, R.; Blazar, B.R. Pathophysiology of Chronic Graft-versus-Host Disease and Therapeutic Targets. N. Engl. J. Med. 2017, 377, 2565–2579.
- Nguyen, H.D.; Kuril, S.; Bastian, D.; Yu, X.-Z. T-Cell Metabolism in Hematopoietic Cell Transplantation. Front. Immunol. 2018, 9, 176.
- Tijaro-Ovalle, N.M.; Karantanos, T.; Wang, H.-T.; Boussiotis, V.A. Metabolic Targets for Improvement of Allogeneic Hematopoietic Stem Cell Transplantation and Graft-vs.-Host Disease. Front. Immunol. 2019, 10, 295.
- Kumari, R.; Palaniyandi, S.; Hildebrandt, G.C. Metabolic Reprogramming-A New Era How to Prevent and Treat Graft Versus Host Disease After Allogeneic Hematopoietic Stem Cell Transplantation Has Begun. Front. Pharmacol. 2020, 11, 588449.
- Buxbaum, N.P.; Farthing, D.E.; Maglakelidze, N.; Lizak, M.; Merkle, H.; Carpenter, A.C.; Oliver, B.U.; Kapoor, V.; Castro, E.; Swan, G.A.; et al. In vivo kinetics and nonradioactive imaging of rapidly proliferating cells in graft-versus-host disease. JCI Insight 2017, 2, e92851.
- Zeiser, R.; Blazar, B.R. Acute Graft-versus-Host Disease—Biologic Process, Prevention, and Therapy. N. Engl. J. Med. 2017, 377, 2167–2179.
- Hill, G.R.; Betts, B.C.; Tkachev, V.; Kean, L.S.; Blazar, B.R. Current Concepts and Advances in Graft-Versus-Host Disease Immunology. Annu. Rev. Immunol. 2021, 39, 19–49.
- Teshima, T. Th1 and Th17 join forces for acute GVHD. Blood 2011, 118, 4765–4767.
- Riegel, C.; Boeld, T.J.; Doser, K.; Huber, E.; Hoffmann, P.; Edinger, M. Efficient treatment of murine acute GvHD by in vitro expanded donor regulatory T cells. Leukemia 2020, 34, 895–908.
- Koreth, J.; Matsuoka, K.-I.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P.; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and Regulatory T Cells in Graft-versus-Host Disease. N. Engl. J. Med. 2011, 365, 2055–2066.
- Edinger, M.; Hoffmann, P.; Ermann, J.; Drago, K.; Fathman, C.G.; Strober, S.; Negrin, R.S. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat. Med. 2003, 9, 1144–1150.
- Matsuoka, K.; Koreth, J.; Kim, H.T.; Bascug, G.; McDonough, S.; Kawano, Y.; Murase, K.; Cutler, C.; Ho, V.T.; Alyea, E.P.; et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci. Transl. Med. 2013, 5, 179ra143.
- Gerriets, V.A.; Rathmell, J.C. Metabolic pathways in T cell fate and function. Trends Immunol. 2012, 33, 168–173.
- Nguyen, H.D.; Chatterjee, S.; Haarberg, K.M.; Wu, Y.; Bastian, D.; Heinrichs, J.; Fu, J.; Daenthanasanmak, A.; Schutt, S.; Shrestha, S.; et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J. Clin. Investig. 2016, 126, 1337–1352.
- Gatza, E.; Wahl, D.R.; Opipari, A.W.; Sundberg, T.B.; Reddy, P.; Liu, C.; Glick, G.D.; Ferrara, J.L. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci. Transl. Med. 2011, 3, 67ra68.
- Hippen, K.L.; Aguilar, E.G.; Rhee, S.Y.; Bolivar-Wagers, S.; Blazar, B.R. Distinct Regulatory and Effector T Cell Metabolic Demands during Graft-Versus-Host Disease. Trends Immunol. 2020, 41, 77–91.
- Cooke, K.R.; Luznik, L.; Sarantopoulos, S.; Hakim, F.T.; Jagasia, M.; Fowler, D.H.; van den Brink, M.R.M.; Hansen, J.A.; Parkman, R.; Miklos, D.B.; et al. The Biology of Chronic Graft-versus-Host Disease: A Task Force Report from the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease. Biol. Blood Marrow Transplant. 2017, 23, 211–234.
- Kondo, M.; Kojima, S.; Horibe, K.; Kato, K.; Matsuyama, T. Risk factors for chronic graft-versus-host disease after allogeneic stem cell transplantation in children. Bone Marrow Transplant. 2001, 27, 727–730.
- Lazaryan, A.; Weisdorf, D.J.; DeFor, T.; Brunstein, C.G.; MacMillan, M.L.; Bejanyan, N.; Holtan, S.; Blazar, B.R.; Wagner, J.E.; Arora, M. Risk Factors for Acute and Chronic Graft-versus-Host Disease after Allogeneic Hematopoietic Cell Transplantation with Umbilical Cord Blood and Matched Sibling Donors. Biol. Blood Marrow Transplant. 2016, 22, 134–140.
- Boyiadzis, M.; Arora, M.; Klein, J.P.; Hassebroek, A.; Hemmer, M.; Urbano-Ispizua, A.; Antin, J.H.; Bolwell, B.J.; Cahn, J.Y.; Cairo, M.S.; et al. Impact of Chronic Graft-versus-Host Disease on Late Relapse and Survival on 7,489 Patients after Myeloablative Allogeneic Hematopoietic Cell Transplantation for Leukemia. Clin. Cancer Res. 2015, 21, 2020–2028.
- Kato, M.; Kurata, M.; Kanda, J.; Kato, K.; Tomizawa, D.; Kudo, K.; Yoshida, N.; Watanabe, K.; Shimada, H.; Inagaki, J.; et al. Impact of graft-versus-host disease on relapse and survival after allogeneic stem cell transplantation for pediatric leukemia. Bone Marrow Transpl. 2019, 54, 68–75.
- Gooptu, M.; Antin, J.H. GVHD Prophylaxis 2020. Front. Immunol. 2021, 12, 694.
- Hamilton, B.K. Current approaches to prevent and treat GVHD after allogeneic stem cell transplantation. Hematology 2018, 2018, 228–235.
- Bertz, H.; Spyridonidis, A.; Wäsch, R.; Grüllich, C.; Egger, M.; Finke, J. A novel GVHD-prophylaxis with low-dose alemtuzumab in allogeneic sibling or unrelated donor hematopoetic cell transplantation: The feasibility of deescalation. Biol. Blood Marrow Transpl. 2009, 15, 1563–1570.
- Saidu, N.E.B.; Bonini, C.; Dickinson, A.; Grce, M.; Inngjerdingen, M.; Koehl, U.; Toubert, A.; Zeiser, R.; Galimberti, S. New Approaches for the Treatment of Chronic Graft-Versus-Host Disease: Current Status and Future Directions. Front. Immunol. 2020, 11, 2625.
- Kanakry, C.G.; O’Donnell, P.V.; Furlong, T.; Lima, M.J.d.; Wei, W.; Medeot, M.; Mielcarek, M.; Champlin, R.E.; Jones, R.J.; Thall, P.F.; et al. Multi-Institutional Study of Post-Transplantation Cyclophosphamide As Single-Agent Graft-Versus-Host Disease Prophylaxis After Allogeneic Bone Marrow Transplantation Using Myeloablative Busulfan and Fludarabine Conditioning. J. Clin. Oncol. 2014, 32, 3497–3505.
- Martin, P.J.; Rizzo, J.D.; Wingard, J.R.; Ballen, K.; Curtin, P.T.; Cutler, C.; Litzow, M.R.; Nieto, Y.; Savani, B.N.; Schriber, J.R.; et al. First- and second-line systemic treatment of acute graft-versus-host disease: Recommendations of the American Society of Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2012, 18, 1150–1163.
- Inamoto, Y.; Flowers, M.E.; Lee, S.J.; Carpenter, P.A.; Warren, E.H.; Deeg, H.J.; Storb, R.F.; Appelbaum, F.R.; Storer, B.E.; Martin, P.J. Influence of immunosuppressive treatment on risk of recurrent malignancy after allogeneic hematopoietic cell transplantation. Blood 2011, 118, 456–463.
- Axt, L.; Naumann, A.; Toennies, J.; Haen, S.; Vogel, W.; Schneidawind, D.; Wirths, S.; Moehle, R.; Faul, C.; Kanz, L. Retrospective single center analysis of outcome, risk factors and therapy in steroid refractory graft-versus-host disease after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2019, 54, 1805–1814.
- Westin, J.R.; Saliba, R.M.; De Lima, M.; Alousi, A.; Hosing, C.; Qazilbash, M.H.; Khouri, I.F.; Shpall, E.J.; Anderlini, P.; Rondon, G. Steroid-refractory acute GVHD: Predictors and outcomes. Adv. Hematol. 2011, 2011.
- Chang, Y.-J.; Zhao, X.-Y.; Huang, X.-J. Strategies for Enhancing and Preserving Anti-leukemia Effects Without Aggravating Graft-Versus-Host Disease. Front. Immunol. 2018, 9, 3041.
- Bantug, G.R.; Galluzzi, L.; Kroemer, G.; Hess, C. The spectrum of T cell metabolism in health and disease. Nat. Rev. Immunol. 2018, 18, 19–34.
- Finlay, D.K. Regulation of glucose metabolism in T cells: New insight into the role of Phosphoinositide 3-kinases. Front. Immunol. 2012, 3, 247.
- Patsoukis, N.; Bardhan, K.; Weaver, J.; Herbel, C.; Seth, P.; Li, L.; Boussiotis, V.A. The role of metabolic reprogramming in T cell fate and function. Curr. Trends Immunol. 2016, 17, 1–12.
- Wang, R.; Green, D.R. Metabolic reprogramming and metabolic dependency in T cells. Immunol. Rev. 2012, 249, 14–26.
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251.
- van der Windt, G.J.W.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42.
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565.
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806.
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338.
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. (Baltim. Md. 1950) 2008, 180, 4476–4486.
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72.
- Mohamed, F.A.; Thangavelu, G.; Rhee, S.Y.; Sage, P.T.; O’Connor, R.S.; Rathmell, J.C.; Blazar, B.R. Recent Metabolic Advances for Preventing and Treating Acute and Chronic Graft Versus Host Disease. Front. Immunol. 2021, 12, 4012.
- Brown, R.A.; Byersdorfer, C.A. Metabolic Pathways in Alloreactive T Cells. Front. Immunol. 2020, 11, 1517.
- Wahl, D.R.; Byersdorfer, C.A.; Ferrara, J.L.M.; Opipari, A.W., Jr.; Glick, G.D. Distinct metabolic programs in activated T cells: Opportunities for selective immunomodulation. Immunol. Rev. 2012, 249, 104–115.
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784.
- Yao, Y.; Wang, L.; Zhou, J.; Zhang, X. HIF-1α inhibitor echinomycin reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. J. Transl. Med. 2017, 15, 28.
- Assmann, J.C.; Farthing, D.E.; Saito, K.; Maglakelidze, N.; Oliver, B.; Warrick, K.A.; Sourbier, C.; Ricketts, C.J.; Meyer, T.J.; Pavletic, S.Z.; et al. Glycolytic metabolism of pathogenic T cells enables early detection of GVHD by 13C-MRI. Blood 2021, 137, 126–137.
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234.
- Zhao, Q.; Chu, Z.; Zhu, L.; Yang, T.; Wang, P.; Liu, F.; Huang, Y.; Zhang, F.; Zhang, X.; Ding, W.; et al. 2-Deoxy-d-Glucose Treatment Decreases Anti-inflammatory M2 Macrophage Polarization in Mice with Tumor and Allergic Airway Inflammation. Front. Immunol. 2017, 8, 637.
- Jalota, A.; Kumar, M.; Das, B.C.; Yadav, A.K.; Chosdol, K.; Sinha, S. Synergistic increase in efficacy of a combination of 2-deoxy-D-glucose and cisplatin in normoxia and hypoxia: Switch from autophagy to apoptosis. Tumor Biol. 2016, 37, 12347–12358.
- Bizjak, M.; Malavašič, P.; Dolinar, K.; Pohar, J.; Pirkmajer, S.; Pavlin, M. Combined treatment with Metformin and 2-deoxy glucose induces detachment of viable MDA-MB-231 breast cancer cells in vitro. Sci. Rep. 2017, 7, 1–14.
- Lu, L.; Chen, Y.; Zhu, Y. The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget 2017, 8, 62793–62802.
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120.
- Feng, Y.; Wu, L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem. Biophys. Res. Commun. 2017, 483, 897–903.
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A.; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; et al. Targeting 6-Phosphofructo-2-Kinase (PFKFB3) as a Therapeutic Strategy against Cancer. Mol. Cancer Ther. 2013, 12, 1461–1470.
- Telang, S.; Clem, B.F.; Klarer, A.C.; Clem, A.L.; Trent, J.O.; Bucala, R.; Chesney, J. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. J. Transl. Med. 2012, 10, 95.
- Salmond, R.J. mTOR Regulation of Glycolytic Metabolism in T Cells. Front. Cell Dev. Biol. 2018, 6, 122.
- Wolfson, R.L.; Sabatini, D.M. The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab. 2017, 26, 301–309.
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPK α1: A glucose sensor that controls CD 8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896.
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844.
- Toschi, A.; Lee, E.; Gadir, N.; Ohh, M.; Foster, D.A. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J. Biol. Chem. 2008, 283, 34495–34499.
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A. mTOR-and HIF-1α–mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 6204.
- Raha, S.; Raud, B.; Oberdörfer, L.; Castro, C.N.; Schreder, A.; Freitag, J.; Longerich, T.; Lochner, M.; Sparwasser, T.; Berod, L.; et al. Disruption of de novo fatty acid synthesis via acetyl-CoA carboxylase 1 inhibition prevents acute graft-versus-host disease. Eur. J. Immunol. 2016, 46, 2233–2238.
- Byersdorfer, C.A.; Tkachev, V.; Opipari, A.W.; Goodell, S.; Swanson, J.; Sandquist, S.; Glick, G.D.; Ferrara, J.L. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood 2013, 122, 3230–3237.
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800.
- Park, M.J.; Lee, S.Y.; Moon, S.J.; Son, H.J.; Lee, S.H.; Kim, E.K.; Byun, J.K.; Shin, D.Y.; Park, S.H.; Yang, C.W.; et al. Metformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between T helper 17 and Tregs. Transl. Res. 2016, 173, 115–130.
- Lee, S.K.; Park, M.-J.; Jhun, J.Y.; Beak, J.-A.; Choi, J.W.; Rye, J.-Y.; Jang, J.W.; Bae, S.H.; Yoon, S.K.; Choi, H.J.; et al. Combination Treatment With Metformin and Tacrolimus Improves Systemic Immune Cellular Homeostasis by Modulating Treg and Th17 Imbalance. Front. Immunol. 2021, 11, 581728.
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014, 20, 953–966.
- Moon, J.; Lee, S.Y.; Choi, J.W.; Lee, A.R.; Yoo, J.H.; Moon, S.J.; Park, S.H.; Cho, M.L. Metformin ameliorates scleroderma via inhibiting Th17 cells and reducing mTOR-STAT3 signaling in skin fibroblasts. J. Transl. Med. 2021, 19, 192.
- Lee, S.Y.; Lee, S.H.; Yang, E.J.; Kim, E.K.; Kim, J.K.; Shin, D.Y.; Cho, M.L. Metformin Ameliorates Inflammatory Bowel Disease by Suppression of the STAT3 Signaling Pathway and Regulation of the between Th17/Treg Balance. PLoS ONE 2015, 10, e0135858.
- Yin, Y.; Choi, S.C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra218.
- Lepez, A.; Pirnay, T.; Denanglaire, S.; Perez-Morga, D.; Vermeersch, M.; Leo, O.; Andris, F. Long-term T cell fitness and proliferation is driven by AMPK-dependent regulation of reactive oxygen species. Sci. Rep. 2020, 10, 21673.
- MacIver, N.J.; Blagih, J.; Saucillo, D.C.; Tonelli, L.; Griss, T.; Rathmell, J.C.; Jones, R.G. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J. Immunol. 2011, 187, 4187–4198.
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516.
- Glick, G.D.; Rossignol, R.; Lyssiotis, C.A.; Wahl, D.; Lesch, C.; Sanchez, B.; Liu, X.; Hao, L.-Y.; Taylor, C.; Hurd, A.; et al. Anaplerotic Metabolism of Alloreactive T Cells Provides a Metabolic Approach To Treat Graft-Versus-Host Disease. J. Pharmacol. Exp. Ther. 2014, 351, 298–307.
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e1719.
- Hong, Y.Q.; Wan, B.; Li, X.F. Macrophage regulation of graft-vs-host disease. World J. Clin. Cases 2020, 8, 1793–1805.
- Chakraverty, R.; Sykes, M. The role of antigen-presenting cells in triggering graft-versus-host disease and graft-versus-leukemia. Blood 2007, 110, 9–17.
- Gaudino, S.J.; Kumar, P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front. Immunol. 2019, 10, 360.
- Reddy, P.; Maeda, Y.; Liu, C.; Krijanovski, O.I.; Korngold, R.; Ferrara, J.L. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat. Med. 2005, 11, 1244–1249.
- Worbs, T.; Hammerschmidt, S.I.; Foerster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 2017, 17, 30–48.
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2019, 9, 3176.
- Cerboni, S.; Gentili, M.; Manel, N. Diversity of pathogen sensors in dendritic cells. Adv. Immunol. 2013, 120, 211–237.
- Amsen, D.; Blander, J.M.; Lee, G.R.; Tanigaki, K.; Honjo, T.; Flavell, R.A. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell 2004, 117, 515–526.
- Kadowaki, N. Dendritic Cells—A Conductor of T Cell Differentiation—. Allergol. Int. 2007, 56, 193–199.
- Zhang, Y.; Louboutin, J.P.; Zhu, J.; Rivera, A.J.; Emerson, S.G. Preterminal host dendritic cells in irradiated mice prime CD8+ T cell-mediated acute graft-versus-host disease. J. Clin. Investig. 2002, 109, 1335–1344.
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749.
- Basit, F.; Mathan, T.; Sancho, D.; de Vries, I.J.M. Human Dendritic Cell Subsets Undergo Distinct Metabolic Reprogramming for Immune Response. Front. Immunol. 2018, 9, 2489.
- Lawless, S.J.; Kedia-Mehta, N.; Walls, J.F.; McGarrigle, R.; Convery, O.; Sinclair, L.V.; Navarro, M.N.; Murray, J.; Finlay, D.K. Glucose represses dendritic cell-induced T cell responses. Nat. Commun. 2017, 8, 15620.
- Elze, M.C.; Ciocarlie, O.; Heinze, A.; Kloess, S.; Gardlowski, T.; Esser, R.; Klingebiel, T.; Bader, P.; Huenecke, S.; Serban, M.; et al. Dendritic cell reconstitution is associated with relapse-free survival and acute GVHD severity in children after allogeneic stem cell transplantation. Bone Marrow Transplant. 2015, 50, 266–273.
- Molina, M.S.; Stokes, J.; Hoffman, E.A.; Eremija, J.; Zeng, Y.; Simpson, R.J.; Katsanis, E. Bendamustine Conditioning Skews Murine Host DCs Toward Pre-cDC1s and Reduces GvHD Independently of Batf3. Front. Immunol. 2020, 11, 1410.
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462.
- Geeraerts, X.; Bolli, E.; Fendt, S.M.; Van Ginderachter, J.A. Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity. Front. Immunol. 2017, 8, 289.
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173.
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430.
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698.
- Joshi, S.; Singh, A.R.; Zulcic, M.; Durden, D.L. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1α and HIF2α stability and tumor growth, angiogenesis, and metastasis. Mol. Cancer Res. 2014, 12, 1520–1531.
- Qualls, J.E.; Subramanian, C.; Rafi, W.; Smith, A.M.; Balouzian, L.; DeFreitas, A.A.; Shirey, K.A.; Reutterer, B.; Kernbauer, E.; Stockinger, S. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe 2012, 12, 313–323.
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24.
- Terakura, S.; Martin, P.; Shulman, H.; Storer, B. Cutaneous macrophage infiltration in acute GvHD. Bone Marrow Transplant. 2015, 50, 1135–1137.
- Piérard, G.; Hermanns-Lê, T.; Paquet, P.; Rousseau, A.-F.; Delvenne, P.; Piérard-Franchimont, C. Toxic epidermal necrolysis and graft-versus-host reaction: Revisiting a puzzling similarity. Int. Sch. Res. Not. 2013, 2013.
- Liu, X.; Su, Y.; Sun, X.; Fu, H.; Huang, Q.; Chen, Q.; Mo, X.; Lv, M.; Kong, Y.; Xu, L.; et al. Arsenic trioxide alleviates acute graft-versus-host disease by modulating macrophage polarization. Sci. China Life Sci. 2020, 63, 1744–1754.
- Seno, K.; Yasunaga, M.; Kajiya, H.; Izaki-Hagio, K.; Morita, H.; Yoneda, M.; Hirofuji, T.; Ohno, J. Dynamics of M1 macrophages in oral mucosal lesions during the development of acute graft-versus-host disease in rats. Clin. Exp. Immunol. 2017, 190, 315–327.
- Sundarasetty, B.; Volk, V.; Theobald, S.J.; Rittinghausen, S.; Schaudien, D.; Neuhaus, V.; Figueiredo, C.; Schneider, A.; Gerasch, L.; Mucci, A. Human effector memory T helper cells engage with mouse macrophages and cause graft-versus-host–like pathology in skin of humanized mice used in a nonclinical immunization study. Am. J. Pathol. 2017, 187, 1380–1398.
- Shin, H.-J.; Baker, J.; Leveson-Gower, D.; Sega, E.I.; Negrin, R. Rapamycin and IL-2 Prevents Lethal Acute Graft-Versus Host Disease by Expansion of Donor Type CD4+CD25+Foxp3+ Regulatory T Cells. Blood 2009, 114, 1334.
- Zeiser, R.; Nguyen, V.H.; Beilhack, A.; Buess, M.; Schulz, S.; Baker, J.; Contag, C.H.; Negrin, R.S. Inhibition of CD4+CD25+ regulatory T-cell function by calcineurin-dependent interleukin-2 production. Blood 2006, 108, 390–399.
- Paz, K.; Flynn, R.; Du, J.; Tannheimer, S.; Johnson, A.J.; Dong, S.; Stark, A.K.; Okkenhaug, K.; Panoskaltsis-Mortari, A.; Sage, P.T.; et al. Targeting PI3Kδ function for amelioration of murine chronic graft-versus-host disease. Am. J. Transplant. 2019, 19, 1820–1830.
- Bronte, V.; Brandau, S.; Chen, S.; Colombo, M.; Frey, A.; Greten, T.; Mandruzzato, S.; Murray, P.; Ochoa, A.; Ostrand-Rosenberg, S. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150.
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8.
- Koehn, B.H.; Apostolova, P.; Haverkamp, J.M.; Miller, J.S.; McCullar, V.; Tolar, J.; Munn, D.H.; Murphy, W.J.; Brickey, W.J.; Serody, J.S. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood J. Am. Soc. Hematol. 2015, 126, 1621–1628.
- Koehn, B.H.; Blazar, B.R. Role of myeloid-derived suppressor cells in allogeneic hematopoietic cell transplantation. J. Leukoc. Biol. 2017, 102, 335–341.
- Koehn, B.H.; Saha, A.; McDonald-Hyman, C.; Loschi, M.; Thangavelu, G.; Ma, L.; Zaiken, M.; Dysthe, J.; Krepps, W.; Panthera, J.; et al. Danger-associated extracellular ATP counters MDSC therapeutic efficacy in acute GVHD. Blood 2019, 134, 1670–1682.
- Cyster, J.G.; Allen, C.D.C. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540.
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971.
- Allen, J.L.; Fore, M.S.; Wooten, J.; Roehrs, P.A.; Bhuiya, N.S.; Hoffert, T.; Sharf, A.; Deal, A.M.; Armistead, P.; Coghill, J.; et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood 2012, 120, 2529–2536.
- McManigle, W.; Youssef, A.; Sarantopoulos, S. B cells in chronic graft-versus-host disease. Hum. Immunol. 2019, 80, 393–399.
- Shimabukuro-Vornhagen, A.; Hallek, M.J.; Storb, R.F.; von Bergwelt-Baildon, M.S. The role of B cells in the pathogenesis of graft-versus-host disease. Blood 2009, 114, 4919–4927.
- Edry, E.; Melamed, D. Receptor editing in positive and negative selection of B lymphopoiesis. J. Immunol. 2004, 173, 4265–4271.
- Smulski, C.R.; Eibel, H. BAFF and BAFF-Receptor in B Cell Selection and Survival. Front. Immunol. 2018, 9, 2285.
- Lesley, R.; Xu, Y.; Kalled, S.L.; Hess, D.M.; Schwab, S.R.; Shu, H.-B.; Cyster, J.G. Reduced Competitiveness of Autoantigen-Engaged B Cells due to Increased Dependence on BAFF. Immunity 2004, 20, 441–453.
- Thien, M.; Phan, T.G.; Gardam, S.; Amesbury, M.; Basten, A.; Mackay, F.; Brink, R. Excess BAFF Rescues Self-Reactive B Cells from Peripheral Deletion and Allows Them to Enter Forbidden Follicular and Marginal Zone Niches. Immunity 2004, 20, 785–798.
- Sarantopoulos, S.; Stevenson, K.E.; Kim, H.T.; Cutler, C.S.; Bhuiya, N.S.; Schowalter, M.; Ho, V.T.; Alyea, E.P.; Koreth, J.; Blazar, B.R.; et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood 2009, 113, 3865–3874.
- Jia, W.; Poe, J.C.; Su, H.; Anand, S.; Matsushima, G.K.; Rathmell, J.C.; Maillard, I.; Radojcic, V.; Imai, K.; Reyes, N.J.; et al. BAFF promotes heightened BCR responsiveness and manifestations of chronic GVHD after allogeneic stem cell transplantation. Blood 2021, 137, 2544–2557.
- Cutler, C.; Miklos, D.; Kim, H.T.; Treister, N.; Woo, S.B.; Bienfang, D.; Klickstein, L.B.; Levin, J.; Miller, K.; Reynolds, C.; et al. Rituximab for steroid-refractory chronic graft-versus-host disease. Blood 2006, 108, 756–762.
- Solomon, S.R.; Sizemore, C.A.; Ridgeway, M.; Zhang, X.; Brown, S.; Holland, H.K.; Morris, L.E.; Solh, M.; Bashey, A. Safety and efficacy of rituximab-based first line treatment of chronic GVHD. Bone Marrow Transplant. 2019, 54, 1218–1226.
- Malard, F.; Labopin, M.; Yakoub-Agha, I.; Chantepie, S.; Guillaume, T.; Blaise, D.; Tabrizi, R.; Magro, L.; Vanhove, B.; Blancho, G.; et al. Rituximab-based first-line treatment of cGVHD after allogeneic SCT: Results of a phase 2 study. Blood 2017, 130, 2186–2195.
- Fowler, D.H.; Pavletic, S.Z. Syk and tired of current chronic GVHD therapies. Blood 2015, 125, 3974–3975.
- Jaglowski, S.M.; Blazar, B.R. How ibrutinib, a B-cell malignancy drug, became an FDA-approved second-line therapy for steroid-resistant chronic GVHD. Blood Adv. 2018, 2, 2012–2019.
- Dubovsky, J.A.; Flynn, R.; Du, J.; Harrington, B.K.; Zhong, Y.; Kaffenberger, B.; Yang, C.; Towns, W.H.; Lehman, A.; Johnson, A.J.; et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J. Clin. Investig. 2014, 124, 4867–4876.
- Schutt, S.D.; Fu, J.; Nguyen, H.; Bastian, D.; Heinrichs, J.; Wu, Y.; Liu, C.; McDonald, D.G.; Pidala, J.; Yu, X.Z. Inhibition of BTK and ITK with Ibrutinib Is Effective in the Prevention of Chronic Graft-versus-Host Disease in Mice. PLoS ONE 2015, 10, e0137641.
- Miklos, D.; Cutler, C.S.; Arora, M.; Waller, E.K.; Jagasia, M.; Pusic, I.; Flowers, M.E.; Logan, A.C.; Nakamura, R.; Blazar, B.R.; et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 2017, 130, 2243–2250.


