 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alan Chiang | + 12926 word(s) | 12926 | 2020-08-06 08:57:34 | | | |
| 2 | Bruce Ren | -6344 word(s) | 6582 | 2020-08-24 07:44:46 | | | | |
| 3 | Bruce Ren | Meta information modification | 6582 | 2020-10-28 03:10:32 | | | | |
| 4 | Bruce Ren | Meta information modification | 6582 | 2020-10-28 03:13:14 | | | | |
| 5 | Bruce Ren | Meta information modification | 6582 | 2020-10-28 03:17:56 | | |
Video Upload Options
Epstein-Barr virus (EBV) lytic induction therapy is an emerging virus-targeted therapeutic approach that exploits the presence of EBV in tumor cells to confer specific killing effects against EBV-associated malignancies. Efforts have been made in the past years to uncover the mechanisms of EBV latent-lytic switch and discover different classes of chemical compounds that can reactivate the EBV lytic cycle. Despite the growing list of compounds showing potential to be used in the lytic induction therapy, only a few are being tested in clinical trials with varying degrees of success. This review will summarize the current knowledge on EBV lytic reactivation, the major hurdles of translating the lytic induction therapy into clinical settings and highlight some potential strategies in the future development of this therapy for EBV-related lymphoid and epithelial malignancies.
1. Introduction
Epstein–Barr virus (EBV) infects more than 90% of adults worldwide. While its primary infection is often asymptomatic, it can manifest as infectious mononucleosis (IM) in adolescents and young adults [1]. EBV is also associated with lymphomas such as endemic Burkitt lymphoma (BL), Hodgkin lymphoma (HL), T-/NK-, and B-cell non-Hodgkin lymphoma as well as epithelial carcinomas, which include undifferentiated nasopharyngeal carcinoma (NPC) and a subset of gastric carcinoma (EBVaGC) [2][3][4]. The biphasic lifecycle of EBV allows it to establish latency subsequent to primary infection in which viral gene expression is limited to those that are responsible for tumorigenesis, apoptosis inhibition, immune evasion, and so on [5]. Owing to the limited choice and the low expression of these viral proteins, it is difficult to target EBV-positive tumor cells specifically. In most cases, treatment against EBV-positive lymphomas is similar to those of EBV-negative lymphomas of the same histology, for example, chemotherapy, radiation, and tumor resection [6]. Therapeutic strategies that target EBV in the associated malignancies can result in highly specific killing effects to the tumor cells, but spare the normal cells from toxic effects.
Occasionally, the latent virus within the infected cells enters into lytic cycle, in which >70 viral proteins are produced [5]. The switch occurs upon the expression of immediate early (IE) proteins, BZLF1 (Zta), and BRLF1 (Rta), which transactivate Zta and Rta promoters (Zp and Rp) and activate the expression of viral genes for viral replication, such as BMRF1, BALF1, and BGLF4, as well as that for production of virions, such as BLLF1 and BFRF3 [7]. The activation of IE proteins and promoters can be achieved through post-translational modification of activators or repressors, modulation of cellular signaling pathways, epigenetic regulation, such as DNA methylation; histone modification; cellular stresses, for example, oxidative stress, hypoxia, autophagy, and inflammation, as well as through modulation of host and viral micro RNAs [8][9][10][11]. Owing to the massive number of viral proteins expressed during the lytic cycle, they may be potentially utilized for EBV-specific therapies. One such therapy is the lytic induction therapy in which EBV is reactivated into the lytic cycle that confers cytotoxicity of antiviral drugs to achieve specific killing effects against EBV-positive cells. Although there were many studies in the past decades studying the lytic induction therapy, only a few were conducted in the setting of clinical trials.
2. Overview of the Lytic Induction Therapy
Lytic induction therapy is an emerging virus-targeted therapeutic approach that exploits the presence of EBV in tumor cells to confer specific killing effects against EBV-associated malignancies. This strategy involves two classes of compounds, that is, chemical lytic inducers and nucleoside analogue antiviral pro-drugs. The EBV lytic cycle is first being reactivated by the chemical lytic inducers producing an array of lytic proteins, one of which is the viral protein kinase encoded by BGLF4 [7]. This kinase phosphorylates and converts nucleoside analog anti-viral pro-drugs, such as ganciclovir, to their cytotoxic forms, consequently killing their host cells. More importantly, the phosphorylated drugs can be transferred to adjacent cells, which leads to a “bystander killing” effect [12] (refer to Figure 1) [13]. As a result, the success of this method relies heavily on the effectiveness of lytic inducers in reactivating EBV lytic cycle, emphasizing the importance of investigating a broad variety of compounds in order to enable and consolidate this form of therapy for EBV-associated malignancies.
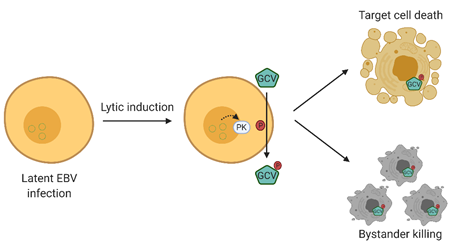
Figure 1. Overview of Epstein–Barr virus (EBV) lytic induction therapy. EBV lytic cycle is first reactivated by chemical inducers in which the viral protein kinase encoded by BGLF4 is produced. BGLF4 then activates the nucleoside analogue antiviral pro-drug into its cytotoxic form, and consequently results in a specific killing effect on EBV-positive cells. Moreover, the activated drugs can be transferred to adjacent cells, resulting in a “bystander killing” effect. GCV, valganciclovir.
3. Lytic Inducers
The lytic induction potential and the modes of lytic reactivation of different compounds, such as histone deacetylase (HDAC) inhibitors, chemotherapeutics agents, phorbol esters, butyrates, and novel compounds, in various cell lines harboring EBV have been summarized in detail in a recent review [14]. Despite having a continuously growing list of lytic inducers that can potentially be incorporated into the lytic induction therapy, very few drugs have been tested in clinical settings [15]. The only clinical trial study to date that has shown a promising outcome tested the effect of combining lytic inducers, gemcitabine (GCb) and valproic acid (VPA), with valganciclovir (GCV) on patients with end-stage NPC [16]. As different classes of lytic inducers have been addressed in detail in other reviews [17][18], we will briefly summarize the mechanisms of EBV lytic reactivation and outline the lytic inducers that possess the corresponding reactivation mechanism.
EBV lytic cycle can be reactivated by modulating different signaling pathways of the host, for example, by activating protein kinase C (PKC) directly or together with mitogen-activated protein kinase (MAPK) family consisting of extracellular-signal-regulated kinase (ERK), c-Jun N-terminal kinases (JNK), and p38 signaling pathways . HDAC inhibitors such as suberanilohydroxamic acid (SAHA), romidepsin, valproic acid (VPA), trichostatin A (TSA), and sodium butyrate (NaB) [19][20]; phorbol esters such as tetradecanoylphorbol acetate (TPA) [21]; and microtubule depolymerization compounds such as colchicine and vinblastine [22] have been shown to activate the PKC and/or JNK and p38 signaling to reactivate EBV lytic cycle. TPA activates nuclear factor-κB (NF-kB) and activator protein 1 (AP-1) that mediate the activation of JNK, which may interact with Zp through the binding of c-Jun to the ZI and ZII elements . Another study revealed that Zp activation via PKC-δ activation requires the ZID element, which allows binding of the transcription factor Sp1 [23]. Proteasome inhibitor such as bortezomib and endoplasmic reticulum (ER) stress inducers such as thapsigargin and tunicamycin, on the other hand, can induce EBV lytic cycle by activating ER stress/unfolded protein response (UPR), which induces JNK and/or C/EBP-β and activates Zp through C/EBP-binding sites in ZII and ZIIIB elements [24][25]. UPR-induced lytic reactivation was also observed in clofoctol treatment, which mediates the activation of the PERK-XBP1 axis [26].
Activation of PI3K/Akt signaling pathway can also reactivate EBV lytic cycle. Compounds that possess this property include chemotherapeutic drugs such as gemcitabine, doxorubicin, cis-platinum, and 5-FU [27] and immunosuppressive drug such as methotrexate [28]. Phosphoinositide 3-kinases (PI3K) activation was shown to be required for Rta activation of Zp and BMRF1 promoters, albeit the exact mechanism has not been completely elucidated [29]. Immunomodulatory agents such as lenalidomide and thalidomide suppress Ikaros, which can regulate EBV latency as well as activate PI3K signaling [30].
Cellular stress-related signaling pathways involving ATM and p53 can also be associated with reactivation of EBV lytic cycle. Reactive oxygen species (ROS) inducers such as H2O2, methylnitronitrosoguanidine (MNNG), and the chemotherapeutic drug gemcitabine activate p53, which subsequently binds to the Sp1-binding element in Zp and Rp and activates the lytic cycle [31][32]. Additionally, chloroquine can reactive EBV lytic cycle by chromatin remodeling through the activation of the ATM pathway and the downstream phosphorylation of KAP1/TRIM28 [33], allowing the access of cellular transcription factors to activate the viral promoters [34].
Induction of hypoxia has been shown to reactivate EBV lytic cycle through the binding of hypoxia-inducible factor 1 (HIF-1) to the hypoxia response element motif on Zp and/or by the activation of the ERK1/2 signaling pathway [35][36]. Iron chelators such as deferoxamine, Dp44mT, and a novel compound known as C7 were found to stabilize HIF-1a, which subsequently leads to the reactivation of EBV lytic cycle [36]. Apart from stabilizing HIF-1a, C7 was also found to reactivate EBV lytic cycle through the activation of the ERK1/2-autophagy (ATG5) axis [36]. In addition to C7, autophagy induction through the PKCδ-p38 MAPK axis by combination of TPA and NaB has also been shown to promote EBV lytic cycle [37].
In addition to the above lytic reactivation pathways, other mechanisms such as induction of psychological stress by glucocorticoids such as hydrocortisone and dexamethasone [38], as well as inhibition of NF-kB signaling by antiretroviral medication such as azidothymidine [39],anti-inflammatory drugs or natural compounds such as aspirin [40] and curcuminoids [41], have been found to reactivate EBV lytic cycle. The detailed mechanisms for the reactivation have not been completely delineated. Large-scale screenings of chemical compounds have also identified several novel organic compounds, named E11 and A10 [42], and tetrahydrocarboline derivatives, named C09, C50, C51, C60, and C67, which can induce EBV lytic induction through as yet undetermined mechanisms [43].
4. Weaknesses and Concerns Related to the Lytic Inducing Compounds
As mentioned in the previous sections, many efforts have been made in the past years to uncover the mechanisms of chemical compounds in reactivating EBV lytic cycle in both EBV-positive lymphomas and epithelial carcinomas. Despite having the potential of being incorporated into EBV lytic induction therapy regimens, as shown in in vitro testing and Phase I/II clinical trial [44], these compounds have major weaknesses in their action. For instance, they have relatively low efficiencies in the reactivation of EBV lytic cycle. Table 1 summarizes the efficiencies of EBV lytic induction by the different compounds from multiple studies. In general, HDAC inhibitors such as NaB could reactivate 2–60% of EBV-positive B cells into lytic cycle, while SAHA could reactivate 30–65% of EBV-associated epithelial cells (AGS-BX1, HA, and HK1-EBV) into lytic cycle [45][46][47]. VPA could induce around 10% of AGS-EBV cells, while the percentage was low in LCL and C666-1 cells [48]. Novel compounds identified by our group such as C7, E11, C8, E7, and A10 could induce 30–60% of AGS-BX1 cells into lytic cycle [42]. Follow-up studies on C7, the best-performing compound identified, showed its ability to induce 6–12% of HA, C666-1, and NPC43 cells into lytic cycle [42]. Another new class of compounds, curcuminoids, were shown to induce 20–50% of AGS-BX1, C666-1, and HONE1-EBV cells into lytic cycle [41]. Combination of lytic compounds such as VPA and cisplatin was able to induce 50% of AGS-EBV cells into lytic cycle [48], while 40–70% could be achieved in AGS-BX1, HONE1-EBV, and C666-1 cells treated with VPA together with gemcitabine [41]. The above studies showed that a considerable proportion of cells are refractory to lytic cycle induction by most compounds studied. This refractory population greatly hinders the implementation of these lytic inducers into the lytic induction therapy and the translation to clinical settings.
Second, these lytic compounds rely heavily on the cellular background for inducing EBV lytic cycle. For example, HDAC inhibitor, VPA, could induce EBV lytic cycle in EBV-associated epithelial carcinomas such as C666-1 and AGS-EBV cells [48], but not in EBV-positive lymphomas such as HH514-16, Raji, and Akata cells [49][50]. NaB was shown to induce lytic cycle in EBV-positive lymphoma cell lines including P3HR-1, B95.8, Raji, Daudi [51], and AK2003 , as well as in EBV-associated epithelial carcinoma cell line, AGS-BX1 , but does so very weakly in NPC cells . SAHA could induce lytic cycle in AGS-BX1, HA , AK2003, and C666-1 cells, but not in NPC43 and LCLs [47]. Similar results were found in chemotherapy agents such as 5-FU and cis-platinum, which could induce lytic cycle in Akata and AGS-EBV cells, but not in LCLs. For other classes of compounds such as tetrahydrocarboline derivatives , curcuminoids, iron chelators, and the novel compounds, lytic induction studies were only examined in either EBV-positive lymphoma cells or a subset of EBV-associated epithelial carcinoma cell lines, thus limiting general conclusions on their abilities to reactivate EBV lytic cycle in both cell types (Table 1). None of the compounds studied to date could induce EBV lytic cycle in all EBV-positive cell lines and their action dependent on cellular background and EBV latency states greatly hinder the incorporation of the available inducers in clinically relevant lytic induction therapeutic regimens.
Lastly, the concern of promoting viral dissemination through chemical induction of EBV lytic cycle has to be addressed with caution. Most of the chemical compounds studied reactivate a complete EBV lytic cycle with production of virions. For instance, supernatant from HONE1-EBV cells induced with SAHA could transduce 71% of Daudi cells in an EBV transduction assay . This raises the concern of promoting viral dissemination in the midst of the therapy. Indeed, a pilot study on the efficacy and safety of romidepsin in treating extranodal natural killer/T-cell lymphoma found a substantial increase in viremia in these patients [52]. The novel compound C7 and anti-bacterial antibiotic, clofoctol, were found to induce the expression of immediately early and early lytic proteins, but not late lytic proteins. Moreover, EBV virions were not produced after lytic induction by these two compounds. The reactivation of EBV lytic cyle without production of virions puts them as potentially suitable candidates for incorporation in lytic induction therapy with minimal risk of viral dissemination.
All of the previously studied compounds have at least one of the three major weaknesses mentioned above. For instance, HDAC inhibitors appear to be efficient in reactivating 30–50% of the cell population into EBV lytic cycle in both EBV-positive lymphoma and epithelial carcinoma cells, but their induction of full viral lytic cycle raises concerns in promoting EBV dissemination during the therapy. On the other hand, C7 and clofoctol are able to induce EBV lytic cycle without production of virions, but a relatively low percentage (10–20%) of cells can be induced into lytic cycle. Therefore, efforts such as structural refinements, as demonstrated in studies by Tikhmyanova et al. [53] and our group , will be important in promoting the utility of these compounds in lytic induction therapy. Apart from refining the currently available lytic inducers, combination of these different classes of compounds, repurposing of other classes of clinically available compounds, or designing novel chemical molecules or peptides can be employed to facilitate the translation of lytic induction therapy for EBV-associated malignancies into the clinics. These strategies will be discussed in detail in section 5.
Table 1. Summary of the efficiency of lytic induction of Epstein–Barr virus (EBV) of the lytic inducers and the cell types in which lytic cycle can be induced *.
|
Class |
Compound |
Cell Type that Can Be Induced (% of Cell Population) |
Cell Type that Cannot Be Induced |
Ref. |
|
HDAC inhibitors |
NaB |
HH514-16, B95.8 |
Raji |
|
|
AHS-BX1, BL-AK2003 |
LCLs |
[47] |
||
|
TSA |
HH514-16, P3J-HR1 |
Raji, B95.8, Akata |
||
|
AHS-BX1, BL-AK2003 |
LCLs |
[47] |
||
|
VPA |
LCL (low), C666-1 (low), AGS-EBV (10%) |
/ |
[48] |
|
|
AGS-BX1 |
LCLs, BL-AK2003 |
[47] |
||
|
TPA |
B95.8, Raji |
HH514-16 |
||
|
SAHA |
AGS-BX1, BL-AK2003 |
LCLs, NPC43 |
[47] |
|
|
HK1-EBV, HONE-1-EBV, HA (30–65%), C666-1 |
/ |
[46] |
||
|
Romidepsin |
HA (75%), C666-1 (6%) |
NPC43 |
[46] |
|
|
DNA methyltransferase inhibitor |
AZC (5 ara2'-deoxycytidine) |
HH514-16 |
/ |
|
|
RaeI (80%) |
/ |
[54] |
||
|
Iron chelators |
Deferoxamine, Dp44mT |
AGS-BX1, SNU719, HA |
/ |
[36] |
|
Deferasirox, Deferiprone |
AGS-BX1, SNU719 |
/ |
[36] |
|
|
Novel compounds |
C7 |
AGS-BX1, SNU719, HONE1-EBV, YCCEL-1, HA (10%), C666-1 (6%), NPC43 (12%) |
/ |
[36] |
|
E11 |
AGS-BX1 (60%), HONE1-EBV, YCCEL-1 |
SNU719, C666-1 |
[42] |
|
|
C8 |
AGS-BX1 (30%), HONE1-EBV, C666-1 |
SNU719, YCCEL-1 |
[42] |
|
|
E7 |
AGS-BX1 (30%), HONE1-EBV, C666-1, SNU719 |
YCCEL-1 |
[42] |
|
|
A10 |
AGS-BX1 (30%), HONE1-EBV, C666-1, YCCEL-1 |
SNU719 |
[42] |
|
|
Chemotherapeutic agents |
5-FU |
Akata, AGS-EBV (24–28%) |
LCL |
[17] |
|
Gemcitabine |
AGS-EBV (30%), Akata, AGS-EBV (24–28%) |
LCL |
[17] |
|
|
Doxorubicin |
LCL [17] |
LCL [48] |
[17] |
|
|
Taxol |
LCL [17] |
LCL [48] |
[17] |
|
|
5 aza-CR |
Akata, AGS-EBV (24–28%) |
/ |
[27] |
|
|
Immunomodulatory agents |
Lenalidomide, thalidomide, pomalidomide |
B95.8, D4 LCL, DAUDI, KEM-I, MUTU-I |
/ |
[17] |
|
Anti-bacterial antibiotic |
Clofoctol |
Akata (40%), SNU719 (2%), C666-1 (0.5%), LCLs (0.5%) |
/ |
[26] |
|
Curcuminoids |
41 |
AGS-BX1 (40–60%), C666-1 (10–30%), HONE1-EBV (20-40%) |
SNU719 |
[41] |
|
EF24 |
AGS-BX1 (50–70%), C666-1 (10–30%), HONE1-EBV (40–60%) |
SNU719 |
[41] |
|
|
Tetrahydrocarboline derivatives |
C09, C50, C53, C60, C67 |
MutuI, LCL, Akata, C666-1 |
/ |
[43] |
|
ER stress inducers |
Thapsigargin |
LCL |
/ |
[25] |
|
ROS inducer |
N-Methyl-N’-Nitro-N-Nitrosoguanidine (MNNG) |
HA, C666-1, NA (70%) |
/ |
[32] |
* HDAC, histone deacetylase; NaB, sodium butyrate; TSA, trichostatin A; VPA, valproic acid; TPA, tetradecanoylphorbol acetate; SAHA, suberanilohydroxamic acid; 5 aza-CR, 5-azacytidine; ER, endoplasmic reticulum; ROS, reactive oxygen species.
5. Potential drugs and strategies in the future development of lytic induction therapy
5.1. Combining currently available lytic inducers for induction of EBV
Different lytic inducers have been combined in previous studies for reactivating lytic cycle of EBV. The combination between TPA and NaB was found to enhance EA-D expression by 1.5-15 fold more than that by either compound alone in Raji cells [55]. Combination of VPA with cisplatin could induce 50% of AGS-EBV cells into lytic cycle with 1.5-5 fold increase relative to treatment with either compound alone. Additionally, when lenalidomide was combined with doxorubicin or melphalan, lytic induction was enhanced in Daudi and Mutu-I cells. These studies showed that combining different classes of lytic inducers with divergent modes of action in lytic reactivation of EBV could complement one another and achieve a higher efficiency in the induction lytic cycle of EBV. Iron chelators and SAHA could reactivate EBV lytic cycle by stabilizing HIF-1a [56] and activating the PKC-d pathway, respectively . Our group showed that iron chelators could reactivate the lytic cycle through autophagy-dependent pathways while SAHA’s action was independent of autophagy. These two compounds might synergize with one another in inducing lytic cycle of EBV (Figure 2a). Combining iron chelators with immunomodulatory agent such as lenalidomide will also be of interest. Lenalidomide could reactivate lytic cycle of EBV by suppressing Ikaros , which is a transcription factor that was found to upregulate the expression of cellular factors responsible for maintaining EBV latency [57][58]. Direct activation of Zta promoter by iron chelator through HIF-1a binding together with the suppression of inhibitory factors that prevent Zta transactivation of other lytic genes by lenalidomide may provide a feed-forward loop for lytic reactivation of EBV (Figure 2b). Some criteria may need to be considered in the design of combination therapy. First, matching the kinetics of lytic induction of different compounds will be important. Our group found that combination of C7 and SAHA could only enhance lytic reactivation when the treatment duration of C7 matched with its reactivation kinetics . Second, compounds of the same class may not utilise the same mode of action in inducing lytic cycle of EBV. VPA antagonized the reactivation of lytic cycle of EBV by other compounds of the same class such as NaB, TSA, AzaCdR, MS-275, apicidin and SAHA and uniquely enhanced expression of some cellular genes . Similar antagonism was also observed when romidepsin, another HDAC inhibitor thought to have similar action as that of SAHA, was combined with C7. Therefore, in-depth study should be performed to delineate the modes of action of different lytic inducers before deciding on the combination therapy.

Figure 2. Combination of currently available lytic inducers. A) Combination of iron chelators and HDAC inhibitors. Iron chelators and HDAC inhibitors reactivate EBV lytic cycle through autophagy-dependent and independent pathways, respectively. Their combination could potentially be synergistic in reactivating EBV lytic cycle. B) Combination of iron chelators and lenalidomide. Combination of lytic inducers with different mechanisms for EBV lytic reactivation may have synergistic effects in reactivating the EBV lytic cycle. Lenalidomide reactivates EBV lytic cycle by suppressing Ikaros, which inhibits the expression of transcription factors that inhibit EBV lytic cycle. Manipulation of Zta expression by iron chelators, together with the suppression of inhibitory factors that prevent Zta transactivation of other lytic genes by lenalidomide, may provide a feed-forward loop for lytic reactivation, thus enhancing EBV lytic induction.
5.2. Repurposing other classes of clinically available compounds for lytic induction of EBV
Many groups have identified new compounds or re-purposed currently available drugs for reactivating lytic cycle of EBV for lytic induction therapy. The following are some other classes of compounds that have not been explored but have been shown to modulate pathways involved in regulating the latent-lytic switch of EBV.
5.2.1. Modulators of autophagy
Autophagy is a conserved cellular mechanism that is involved in regulating cellular homeostasis as well as governing cell death and survival. Its progression involves a number of sequential events ,i.e., vesicle initiation, elongation, maturation, fusion and degradation, that involve many different tightly regulated autophagic proteins [59][60][61]. Several studies have reported the interaction between the autophagy machinery and EBV latent proteins. For instance, the autophagy machinery processed EBNA1 for its presentation on MHC-II molecules in EBV-positive B cells [62]. Expression of autophagic proteins was enhanced in B cells and epithelial cells by EBNA3C [63] and LMP2A [64], respectively. Furthermore, LMP1 was shown to initiate autophagy progression in B cells [65][66]. Apart from EBV latent proteins, Rta was shown to initiate autophagy through ERK1/2 signaling pathway. The same study also found that autophagy inhibition by 3-MA or ATG5 knockdown abrogated the expression of EBV lytic proteins and production of virions in B cells [67]. Therefore, modulators of autophagy may represent a potential new class of compounds to be employed for lytic reactivation of EBV. Indeed, our group showed that C7 and iron chelators could reactivate lytic cycle of EBV through autophagy, in particular, through ATG5-related mechanisms. Furthermore, bafilomycin A1, an inhibitor of autophagy, enhanced expression of EBV lytic genes in Akata and Mutu-I cells[68]. On the other hand, an mTOR inhibitor, rapamycin, that activates autophagy, was found to induce lytic cycle of EBV in EBV-associated epithelial cells [69] but not in B cells [70]. In addition, the effects of other pre-clinical or clinically available modulators of autophagy in the reactivation of lytic cycle of EBV have not been studied in detail. For example, an activator of autophagy, genistein, was shown to have anti-tumor effects in a phase II clinical trial for prostate cancer [71] and might be a potential compound to be investigated for its role in lytic reactivation of EBV [72][73]. New compounds that have more specific action on particular autophagic proteins such as ULK1, Vps34 andATG4B have also been developed [74]. Exploring different modulators of autophagy on their effects in inducing lytic cycle of EBV may be relevant for the development of lytic induction therapy (Figure 3).

Figure 3. Relationship between induction of lytic cycle of EBV and the autophagy machinery and the modes of action of compounds with lytic induction potentials. EBNA1 could be processed by the autophagy machinery for MHC-II presentation while LMP1, LMP2A, Rta and Zta could initiate autophagy. Rapamycin reactivates EBV lytic cycle by inhibiting mTOR. Iron chelators and C7, on the other hand, activate the ERK1/2-ATG5 axis to induce the lytic cycle of EBV. New compounds that target ATG4, ULK1 and Vps34 could potentially reactivate lytic cycle of EBV.
5.2.2. Modulators of NF-kB signaling
NF-kB transcription factors consist of the REL family members, i.e., RelA(p65), RelB, c-Rel, p50, and p52, that are involved in regulating the proliferation, differentiation and survival of lymphoid cells as well as modulating innate and adaptive immune responses [75]. In response to receptor signal transduction, degradation of the NF-kB inhibitor, IkB, results in the translocation of the transcription factor from the cytosol to the nucleus for transcriptional activation of genes including Blimp1 [76], HIF-1a [77] and YY1[78], which are known transcription factorsthat have been shown to modulate lytic cycle of EBV. In addition, activation of NF-kB signaling pathway was shown to be involved in the pathogenesis of EBV-associated diseases. For instance, LMP1 was reported to activate both canonical and non-canonical NF-kB signaling pathways [79][80]. RelA(p65) was shown to bind and activate Qp-EBNA1 expression in EBV-associated epithelial cells [81] while EBNA1 could, in turn, inhibit RelA(p65) by preventing IkK phosphorylation [82]. A more complex interaction was found between Zta and the NF-kB pathway. Interaction between RelA(p65) and Zta abrogated the ability of Zta to transactivate other genes [83][84]. At the same time, Zta inhibited the activation of NF-kB-responsive gene promoters [85], including IkB, which normally retains the inactive NF-kB in the cytoplasm. As a result, high level of NF-kB was observed in the nucleus [86] along with Zta, inhibiting one another. On the other hand, BGLF2 was shown to interact with RelA(p65), preventing its phosphorylation and nuclear translocation [87]. Therefore, inhibiting NF-kB may lead to reactivation of lytic cycle of EBV lytic [88]. Some previously reported lytic inducers also act by modulating the NF-kB pathways. For example, a proteasome inhibitor, bortezomib, could reactivate lytic cycle of EBV in Akata and RaeI cells [24] and inhibit the activation of the NF-kB pathway by preventing the proteasomal degradation of the NF-kB inhibitor, IkBa [89]. Aspirin was shown to reactivate EBV lytic cycle in B95.8 and Raji cells by inhibiting RelA(p65) translocation to the nucleus . Due to the complex interactions among NF-kB, its regulated gene products and EBV proteins such as Zta, detailed investigations of the effects of modulators of NF-kB signaling pathway on the reactivation of lytic cycle of EBV are indicated. Some potential compounds, including a small molecule, known as PS1145, could specifically inhibit IkB phosphorylation and degradation and the subsequent nuclear translocation of NF-kB in NPC cells [90]. It was also reported to induce lytic cycle of EBV in another study [91]. A sesquiterpene lactone, pathenolide, found in medicinal plants such as feverfew could also inhibit NF-kB and activate the expression of Zta and Rta in Raji cells[92]. Therefore, it is of interest to study the effects of modulators of NF-kB signaling pathway in the reactivation of lytic cycle of EBV (Figure 4).
5.2.3 Inhibitors of STAT3
STAT3 is a transcription factor that regulates a number of physiological processes including apoptosis, immune responses and cell proliferation. Cytokine such as IL-6 or engagement of growth factor receptors mediates the activation of STAT3, which subsequently translocates to the nucleus and activates the transcription of genes that are involved in the aforementioned biological processes [93][94]. STAT3 is also closely related to the function of various EBV proteins. For instance, LMP2A was found to induce the phosphorylation of STAT3 that activates DNMT1 transcription and leads to the loss of PTEN expression, a common phenomenon observed in EBV-associated gastric carcinoma [95]. A positive auto-regulatory loop between LMP1 and STAT activation was reported in NPC cells [96]. A subsequent study showed that LMP1 triggered the NF-kB, AP-1 and STAT signaling pathways in NPC cells [97] while NF-kB, AKT and STAT3 were activated by LMP1 in B lymphoma cells [98]. STAT3 was constitutively activated in EBV-positive T or NK lymphoma cell lines [99]. Daigle et al. found that STAT3 level was substantially increased in EBV-positive B cells that were refractory to induction of lytic cycle by NaB [100]. Knockdown of STAT3 sensitized BL cells to lytic inducers for reactivation of lytic cycle while STAT3 inhibition by small molecules, AG490, WP1066 or stattic was found to reactivate lytic cycle of EBV and enhance induction of lytic cycle in EBV-positive BL cells and LCLs by NaB or Aza [101]. Furthermore, icaritin inhibited STAT3 and AKT pathways by downregulating LMP1 expression, which consequently induced EBV lytic gene expression in ENKL cell lines [102]. In contrast, berberine was found to repress the level of EBNA1 by inhibiting EBNA1 promoter Qp. It also inhibits p-STAT3, consequently reducing the expression of EBV lytic genes and production of virions in HONE1 and HK1-EBV cells upon treatment with NaB and TPA [103]. Cucurbitacin I was found to possess anti-proliferative effects in NPC cells by inhibiting STAT3 phosphorylation. However, its effect in reactivation of lytic cycle of EBV was not examined [104]. JAK2/STAT3 inhibitor such as AZD1480 was found to suppress STAT3 without affecting ERK and AKT signaling pathways [105][106]. Other STAT3 inhibitors such as S3I-201 [107], STA-21 [108], 5,15-DPP [109] and S3I-1757 [110], which prevent STAT3 homodimerization, DNA-binding and transcriptional activities, should be investigated in their effects on reactivation of lytic cycle of EBV (Figure 4).

Figure 4. Relationship between EBV proteins, NF-kB and STAT3 signaling pathways and the modes of action of compounds with lytic induction potentials. LMP1 could activate both canonical and non-canonical NF-kB pathways. RelA(p65) could bind and activate Qp-EBNA1 expression while it itself could, in turn, be inhibited by EBNA1 through the prevention of IkK phosphorylation. RelA(p65) interacts with Zta and abrogates its ability to transactivate other EBV genes while Zta inhibits the activation of NF-kB-responsive gene promoters. EBV lytic protein encoded by BGLF2 was shown to interact with RelA(p65), preventing its phosphorylation and nuclear translocation. Bortezomib, PS1145 and aspirin reactivate EBV lytic cycle by preventing the degradation of NF-kB inhibitor, the phosphorylation of Ikβα and translocation of RelA(p65), respectively. Both LMP1 and LMP2A could phosphorylate STAT and inhibit the activation of lytic cycle of EBV. STAT inhibitors such as cucurbitacin I, AZD1480 and S3I-201 reactivate lytic cycle of EBV by either inhibiting phosphorylation, homodimerization, DNA binding or transcriptional activities of STAT3.
5.2.4. Inhibitors of hTERT/NOTCH signaling
EBV proteins promote tumorigenesis through different mechanisms, one of which is by activating the hTERT promoter by LMP1 through NF-kB, MAPK and ERK1/2 signaling pathways in B cells and through c-MYC in NPC cells [111]. hTERT is the catalytic component of telomerase which can stabilize telomeres, preventing it from shortening after rounds of cell cycles, thus contributing to the immortalization of cells [112]. In addition, hTERT inhibits the expression of BZLF1 through the NOTCH2/BATF pathway [113] and hTERT silencing by shRNA induces lytic cycle of EBV in BL and LCLs [114]. Apart from its potential role in lytic reactivation, inhibiting telomerase itself may also inhibit tumorigenesis. It would be of interest to study the lytic reactivation ability and cytotoxic effects of the available hTERT inhibitors on EBV-positive cancers. A hTERT inhibitor, BIBR1532, is a synthetic non-nucleoside compound that can selectively inhibit telomerase activity [115]. It can also induce senescence in human cancer cells [116] and possess anti-proliferative effects to leukemia cells but not normal hematopoietic stem cells [117][118]. It can mediate S-phase arrest in LCLs and BL cells and result in apoptosis [119]. However, its effect on lytic reactivation of EBV has not been studied. Cautions have to be taken with the use of hTERT inhibitors as long term exposure such as 130 days’ treatment of a hTERT inhibitor, MST-312 [120][121][122], was found to cause cell adaptation by overexpression of telomerase in response to the inhibition in breast cancer cells [123]. Moreover, costunolide was shown to diminish hTERT expression as well as EZH2, H3K27me3 and MSH2 levels in glioblastoma cells. Disruption of EZH2 was shown to increase the expression of both EBV lytic and latent genes including LMP1 [124], suggesting that some hTERT inhibitors might cause reveral of the induction of lytic cycle [125][126] and short incubation time with these inhibitors might be warranted.
As hTERT inhibits the expression of BZLF1 through the NOTCH2/BATF pathway , targeting the NOTCH signaling pathway and examining its effects on induction of lytic cycle of EBV will be of interest. NOTCH receptors are located in the plasma membrane. Upon ligand binding, cleavage on different domains of the NOTCH receptor will occur. The intracellular domain (Notch-IC) released from the transmembrane domain by γ-secretase enters the nucleus and interacts with the transcription factor complex that consequently activates a number of NOTCH target genes such as MYC and p21 [127]. In the context of EBV, EBNA2 is regarded as the functional homolog of active Notch-IC [128] while LMP2A can activate the NOTCH pathway [129]. Furthermore, activated NOTCH2 was shown to inhibit the reactivation of lytic cycle of EBV through the upregulation of Zeb2, a transcription factor that represses BZLF1 transcription in B cells [130]. Moreover, γ-secretase inhibitors including compound E and dibenzazepine prevented the cleavage of NOTCH2 and inhibited the release of Notch-IC and could transactivate lytic cycle of EBV in LCLs (please check). Another γ-secretase inhibitor, DAPT, may be able to reactivate the lytic cycle of EBV by reducing the amount of cleaved Notch1-IC and the expression of transcription factors involved in endothelial-mesenchymal transition such as ZEB1 and ZEB2 [131]. Indeed, treatment with doxycycline and DAPT in KSHV-infected iSLK.RGB cells was found to increase mRNA expression of viral lytic genes [132]. L-685,458 was also shown to down-regulate c-MYC expression as well as NF-kB and NOTCH pathways in tongue carcinoma cells[133]. It will be of interest to examine the effects of these inhibitors on the reactivation of lytic cycle of EBV (Figure 5).

Figure 5. Relationship between EBV proteins, hTERT and NOTCH signaling pathway and the modes of action of compounds with lytic induction potentials. hTERT inhibits the expression of BZLF1 through the NOTCH2/BATF pathway. EBNA2 is regarded as the functional homolog of active Notch-IC and LMP2A can activate the NOTCH pathway. NOTCH2 inhibits the reactivation of lytic cycle of EBV through the upregulation of Zeb2 by NOTCH-IC, a transcription factor that represses BZLF1 transcription. γ-secretase inhibitors such as compound E and dibenzazepine can reactivate lytic cycle of EBV by preventing the release of Notch-IC. Other compounds that may reactivate the lytic cycle include hTERT inhibitor, BIBR1532, which selectively inhibits telomerase activity and another γ-secretase inhibitor, DAPT.
5.2.5. Inhibitors of MYC
MYC regulates many different essential cellular processes including cell proliferation, cell-cycle progression, DNA repair and survival. Under normal circumstances, MYC expression is tightly regulated and it has been shown to be deregulated in over 50% of human cancers [134][135]. In the context of EBV, a recent study on identifying host factors that repress lytic cycle of EBV by a human genome-wide CRISPR-Cas9 screen was performed in BL cells. The identified host repressors were found to be centered on MYC. It was found that MYC bound to the OriLyt on the EBV genome and suppressed its looping to the BZLF1 promoter. Furthermore, depletion of MYC or factors related to MYC expression reactivated the lytic cycle, suggesting that MYC inhibition could reactivate EBV lytic cycle [136]. Although MYC inhibition would be a direct and powerful approach for the treatment of many types of cancers, MYC lacks a specific active site for binding by small molecules. Therefore, this “undruggable protein structure” greatly hinders the development of chemical compounds inhibiting MYC [137]. As a result, different compounds that indirectly target MYC such as interrupting MYC transcription [138][139], stability [140][141]and the MYC-MAX complex [142] have been developed. MYC transcription is under the regulation of Bromodomain-containing 4 (BRD4) and a BRD4 inhibitor, JQ1, was found to possess anti-tumor effects. Of interest, JQ1 was found to inhibit the lytic reactivation of EBV as JQ1 not only inhibited MYC expression but also other host factors required for activation of lytic cycle including BACH1, whose knockdown reduced the expression of BZLF1 upon treatment with gemcitabine[143]. Similarly, inhibiting CDK7, a transcription factor that regulates MYC expression [144], prevented EBV replication [145] but significantly inhibited cell growth of NPC [146]. Despite the above observations, it was found that DRB’s inhibition of CDK9, another transcription factor that regulates MYC transcription, reduced both MYC and EBNA2-activated transcription [147]. Since EBNA2 is the functional homologue of NOTCH that inhibits activation of lytic cycle of EBV, inhibiting EBNA2’s function may reverse the inhibition on lytic cycle. Upon initiation of lytic cycle by transfection of BZLF1 in HEK/EBV cells and incubation with CDK2/CDK9 inhibitor or A2CE, only the expression of late lytic proteins but not the early lytic proteins was reduced [148]. Other compounds that target the DNA binding domain of the MYC-MAX complex such as KSI-3716 [149], MYCi975 [150], sAJM589 [151] and 10074-G5 [152][153] were found to have anti-tumor effects in multiple tumor cell types. New compounds such as PKUMDL-YC-1202-1205 [154], 7594-0035[155], VPC70063 [156] and JKY-2-169 [157] have also been developed. It would be of interest to investigate the effects of these compounds on the reactiovation of lytic cycle of EBV (Figure 6).

Figure 6. Relationship between EBV proteins and c-MYC and the modes of action of compounds with lytic induction potentials. MYC represses the activation of lytic cycle of EBV by binding to OriLyt on the EBV genome and suppresses its looping to the BZLF1 promoter. A2CE’s and DRB’s inhibition of CDK2/9, a transcription factor that regulates MYC expression, can suppress the expression of EBV latent proteins. Compounds that target the DNA binding domain of the MYC-MAX complex such as KSI-3716, MYCi975 and 7594-0035 may reactivate the lytic cycle of EBV.
5.3. Designing peptides or small chemical molecules for lytic induction of EBV
Protein structural analyses using NMR, Cryo-EM and crystallography as well as the increasing usage of computer programs in the prediction of active functional domains of protein and docking simulation have accelerated the progress of structure-based drug discovery [158][159][160]. For example, an EBNA1-specific probe was designed and shown to disrupt EBNA1 oligomerization and transactivation. Furthermore, the probe could reactivate lytic cycle of EBV, indicating that inhibition of repressor of lytic cycle can be harnessed to reactivate lytic cycle of EBV [161][162][163][164]. Hence, cellular factors that were shown previously to inhibit reactivation of lytic cycle can be targeted. Examples are Oct-2 and Pax-5, which are B cell-specific transcription factors [165][166] shown to interact directly with Zta and prevent its binding and transactivation of EBV gene promoters. Furthermore, knockdown of either Oct-2 or Pax-5 could increase expression of lytic proteins[167]. In contrast, the cellular factor, NF-Y, was shown to bind to Rp and the overexpression of NF-YA enhanced the expression of Zta and Rta in NPC. Molecules can be designed to stabilize the binding of NF-Y to the IE promoters which may, in turn, enhance reactivation of lytic cycle of EBV [168]. Advancement in computational modelling and the resolution of structural interactions between proteins and compounds or peptides can lead to the rational design of highly specific molecules to be incorporated in lytic induction therapy.
6. Beyond lytic induction therapy
Apart from lytic induction therapy, identification of essential host factors for the survival of EBV-positive cells can be manipulated to facilitate the development of synthetic lethality. For example, BATF and IRF4 were found to be upregulated upon EBV infection of primary B cells. This resulted in the suppression of BIM and upregulation of MYC, which were found to be important transformation factors for primary B cells upon EBV infection. Knockout of either gene triggered apoptosis of EBV-LCL, suggesting that LCL is addicted to BATF and IRF4 for survival [169]. IRF4 antisense oligonucleotides were found to possess anti-tumor activity in multiple myeloma [170], which was also found to be addicted to IRF4 for survival. Recently, a high-throughput screening of chemical compounds that deplete IRF4 identified several compounds of interest [171] which may be relevant in novel therapeutic approaches against EBV-positive lymphomas. Likewise, the consequences on the host cells upon induction of lytic cycle may potentially be manipulated as therapeutic approaches against EBV-associated cancers. For example, many of the EBV lytic proteins, such as BGLF4 (viral protein kinase), BGLF5 (DNA exonuclease), BALF3 (terminase) and BNRF1 (major tegument protein) were found to induce genome instability [172][173][174][175]. Furthermore, BPLF1 (large tegument protein and deubiquitinating (DUB) enzyme) was found to regulate DNA damage response (DDR) by targeting ubiquitinated proliferating cell nuclear antigen (PCNA). Moreover, overexpression of BPLF1 deubiquitinated PCNA, abolished DDR and sensitized EBV-positive cells to ultraviolet light and hydroxyurea [176]. Further inhibition of DNA repair mechanism by chemical drugs may overload EBV-positive cells that undergo lytic cycle to DNA damage thus killing the cells. Advancement of omics technologies may serve to provide an overview of the virus-host interactions and identify host factors that regulate the lytic cycle, which eventually leads to new directions in the design of therapeutic strategies against EBV-associted malignancies.
7. Conclusions
In this review, we have summarized the current knowledge of the reactivation of lytic cycle of EBV and the lytic inducers which have been studied in the past decades. We have also addressed the three major weaknesses of the lytic induction therapy, namely, the relatively low efficiency and the high reliance on the cellular background of lytic inducers in the lytic reactivation of EBV and the concern of viral dissemination during lytic induction therapy. In addition, we have suggested future strategies such as combining different classes of lytic inducing compounds, repurposing other classes of clinically available compounds or designing novel chemical molecules or peptides to potentiate and translate lytic induction therapy into the clinical settings. Identification of EBV-dependent host factors and proteins involved in the reactivation of lytic cycle will expand our basic understanding of EBV biology and provide valuable insights in the development of new therapeutic approaches against EBV-associated malignancies.
References
- Odumade, O.A.; Hogquist, K.A.; Balfour, H.H., Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin. Microbiol. Rev. 2011, 24, 193–209, doi:10.1128/CMR.00044-10.
- Niedobitek, G.; Meru, N.; Delecluse, H.J. Epstein-Barr virus infection and human malignancies. Int J. Exp. Pathol 2001, 82, 149–170, doi:10.1046/j.1365-2613.2001.iep0082-0149-x.
- Delecluse, H.J.; Feederle, R.; O'Sullivan, B.; Taniere, P. Epstein Barr virus-associated tumours: An update for the attention of the working pathologist. J. Clin. Pathol. 2007, 60, 1358–1364, doi:10.1136/jcp.2006.044586.
- Hui, K.F.; Chan, T.F.; Yang, W.; Shen, J.J.; Lam, K.P.; Kwok, H.; Sham, P.C.; Tsao, S.W.; Kwong, D.L.; Lung, M.L., et al. High risk Epstein-Barr virus variants characterized by distinct polymorphisms in the EBER locus are strongly associated with nasopharyngeal carcinoma. Int. J. Cancer 2019, 144, 3031–3042, doi:10.1002/ijc.32049.
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337, doi:10.1056/NEJMra032015.
- Kanakry, J.A.; Ambinder, R.F. EBV-related lymphomas: New approaches to treatment. Curr. Treat. Options Oncol. 2013, 14, 224–236, doi:10.1007/s11864-013-0231-y.
- Israel, B.F.; Kenney, S.C. Virally targeted therapies for EBV-associated malignancies. Oncogene 2003, 22, 5122–5130, doi:10.1038/sj.onc.1206548.
- Li, H.; Liu, S.; Hu, J.; Luo, X.; Li, N.; A, M.B.; Cao, Y. Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int. J. Biol Sci. 2016, 12, 1309–1318, doi:10.7150/ijbs.16564.
- Lin, Z.; Wang, X.; Fewell, C.; Cameron, J.; Yin, Q.; Flemington, E.K. Differential expression of the miR-200 family microRNAs in epithelial and B cells and regulation of Epstein-Barr virus reactivation by the miR-200 family member miR-429. J. Virol. 2010, 84, 7892–7897, doi:10.1128/JVI.00379-10.
- Ellis-Connell, A.L.; Iempridee, T.; Xu, I.; Mertz, J.E. Cellular microRNAs 200b and 429 regulate the Epstein-Barr virus switch between latency and lytic replication. J. Virol. 2010, 84, 10329–10343, doi:10.1128/JVI.00923-10.
- Chen, Y.; Fachko, D.; Ivanov, N.S.; Skinner, C.M.; Skalsky, R.L. Epstein-Barr virus microRNAs regulate B cell receptor signal transduction and lytic reactivation. PLoS Pathog. 2019, 15, e1007535, doi:10.1371/journal.ppat.1007535.
- Freeman, S.M.; Abboud, C.N.; Whartenby, K.A.; Packman, C.H.; Koeplin, D.S.; Moolten, F.L.; Abraham, G.N. The "bystander effect": Tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993, 53, 5274–5283.
- Meng, Q.; Hagemeier, S.R.; Fingeroth, J.D.; Gershburg, E.; Pagano, J.S.; Kenney, S.C. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol. 2010, 84, 4534–4542, doi:10.1128/JVI.02487-09.
- Hui, K.F.; Yiu, S.P.T.; Tam, K.P.; Chiang, A.K.S. Viral-Targeted Strategies Against EBV-Associated Lymphoproliferative Diseases. Front. Oncol. 2019, 9, 81, doi:10.3389/fonc.2019.00081.
- Hutajulu, S.H.; Kurnianda, J.; Tan, I.B.; Middeldorp, J.M. Therapeutic implications of Epstein-Barr virus infection for the treatment of nasopharyngeal carcinoma. Ther. Clin. Risk Manag. 2014, 10, 721–736, doi:10.2147/TCRM.S47434.
- Wildeman, M.A.; Novalic, Z.; Verkuijlen, S.A.; Juwana, H.; Huitema, A.D.; Tan, I.B.; Middeldorp, J.M.; de Boer, J.P.; Greijer, A.E. Cytolytic virus activation therapy for Epstein-Barr virus-driven tumors. Clin. Cancer Res. 2012, 18, 5061–5070, doi:10.1158/1078-0432.CCR-12-0574.
- Feng, W.H.; Hong, G.; Delecluse, H.J.; Kenney, S.C. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. J. Virol. 2004, 78, 1893–1902.
- Li, H.; Hu, J.; Luo, X.; Bode, A.M.; Dong, Z.; Cao, Y. Therapies based on targeting Epstein-Barr virus lytic replication for EBV-associated malignancies. Cancer Sci. 2018, 109, 2101–2108, doi:10.1111/cas.13634.
- Hui, K.F.; Cheung, A.K.; Choi, C.K.; Yeung, P.L.; Middeldorp, J.M.; Lung, M.L.; Tsao, S.W.; Chiang, A.K. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int. J. Cancer 2016, 138, 125–136, doi:10.1002/ijc.29698.
- Lee, H.H.; Chang, S.S.; Lin, S.J.; Chua, H.H.; Tsai, T.J.; Tsai, K.; Lo, Y.C.; Chen, H.C.; Tsai, C.H. Essential role of PKCdelta in histone deacetylase inhibitor-induced Epstein-Barr virus reactivation in nasopharyngeal carcinoma cells. J. Gen. Virol. 2008, 89, 878–883, doi:10.1099/vir.0.83533-0.
- Gao, X.; Ikuta, K.; Tajima, M.; Sairenji, T. 12-O-tetradecanoylphorbol-13-acetate induces Epstein-Barr virus reactivation via NF-kappaB and AP-1 as regulated by protein kinase C and mitogen-activated protein kinase. Virology 2001, 286, 91–99, doi:10.1006/viro.2001.0965.
- Liu, Y.R.; Huang, S.Y.; Chen, J.Y.; Wang, L.H. Microtubule depolymerization activates the Epstein-Barr virus lytic cycle through protein kinase C pathways in nasopharyngeal carcinoma cells. J. Gen. Virol. 2013, 94, 2750–2758, doi:10.1099/vir.0.058040-0.
- Tsai, P.F.; Lin, S.J.; Weng, P.L.; Tsai, S.C.; Lin, J.H.; Chou, Y.C.; Tsai, C.H. Interplay between PKCdelta and Sp1 on histone deacetylase inhibitor-mediated Epstein-Barr virus reactivation. J. Virol. 2011, 85, 2373–2385, doi:10.1128/JVI.01602-10.
- Shirley, C.M.; Chen, J.; Shamay, M.; Li, H.; Zahnow, C.A.; Hayward, S.D.; Ambinder, R.F. Bortezomib induction of C/EBPbeta mediates Epstein-Barr virus lytic activation in Burkitt lymphoma. Blood 2011, 117, 6297–6303, doi:10.1182/blood-2011-01-332379.
- Taylor, G.M.; Raghuwanshi, S.K.; Rowe, D.T.; Wadowsky, R.M.; Rosendorff, A. Endoplasmic reticulum stress causes EBV lytic replication. Blood 2011, 118, 5528–5539, doi:10.1182/blood-2011-04-347112.
- Lee, J.; Kosowicz, J.G.; Hayward, S.D.; Desai, P.; Stone, J.; Lee, J.M.; Liu, J.O.; Ambinder, R.F. Pharmacologic Activation of Lytic Epstein-Barr Virus Gene Expression without Virion Production. J. Virol. 2019, 93, doi:10.1128/JVI.00998-19.
- Feng, W.H.; Israel, B.; Raab-Traub, N.; Busson, P.; Kenney, S.C. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Res. 2002, 62, 1920–1926.
- Feng, W.H.; Cohen, J.I.; Fischer, S.; Li, L.; Sneller, M.; Goldbach-Mansky, R.; Raab-Traub, N.; Delecluse, H.J.; Kenney, S.C. Reactivation of latent Epstein-Barr virus by methotrexate: A potential contributor to methotrexate-associated lymphomas. J. Natl. Cancer Inst. 2004, 96, 1691–1702, doi:10.1093/jnci/djh313.
- Darr, C.D.; Mauser, A.; Kenney, S. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. J. Virol. 2001, 75, 6135–6142, doi:10.1128/JVI.75.13.6135-6142.2001.
- Jones, R.J.; Iempridee, T.; Wang, X.; Lee, H.C.; Mertz, J.E.; Kenney, S.C.; Lin, H.C.; Baladandayuthapani, V.; Dawson, C.W.; Shah, J.J., et al. Lenalidomide, Thalidomide, and Pomalidomide Reactivate the Epstein-Barr Virus Lytic Cycle through Phosphoinositide 3-Kinase Signaling and Ikaros Expression. Clin. Cancer Res. 2016, 22, 4901–4912, doi:10.1158/1078-0432.CCR-15-2242.
- Lee, H.G.; Kim, H.; Kim, E.J.; Park, P.G.; Dong, S.M.; Choi, T.H.; Kim, H.; Chong, C.R.; Liu, J.O.; Chen, J., et al. Targeted therapy for Epstein-Barr virus-associated gastric carcinoma using low-dose gemcitabine-induced lytic activation. Oncotarget 2015, 6, 31018–31029, doi:10.18632/oncotarget.5041.
- Huang, S.Y.; Fang, C.Y.; Wu, C.C.; Tsai, C.H.; Lin, S.F.; Chen, J.Y. Reactive oxygen species mediate Epstein-Barr virus reactivation by N-methyl-N'-nitro-N-nitrosoguanidine. PLoS ONE 2013, 8, e84919, doi:10.1371/journal.pone.0084919.
- Li, X.; Burton, E.M.; Bhaduri-McIntosh, S. Chloroquine triggers Epstein-Barr virus replication through phosphorylation of KAP1/TRIM28 in Burkitt lymphoma cells. PLoS Pathog. 2017, 13, e1006249, doi:10.1371/journal.ppat.1006249.
- Ramasubramanyan, S.; Osborn, K.; Flower, K.; Sinclair, A.J. Dynamic chromatin environment of key lytic cycle regulatory regions of the Epstein-Barr virus genome. J. Virol. 2012, 86, 1809–1819, doi:10.1128/JVI.06334-11.
- Kraus, R.J.; Yu, X.; Cordes, B.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z., et al. Hypoxia-inducible factor-1alpha plays roles in Epstein-Barr virus's natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404, doi:10.1371/journal.ppat.1006404.
- Yiu, S.P.T.; Hui, K.F.; Choi, C.K.; Kao, R.Y.T.; Ma, C.W.; Yang, D.; Chiang, A.K.S. Intracellular Iron Chelation by a Novel Compound, C7, Reactivates Epstein(-)Barr Virus (EBV) Lytic Cycle via the ERK-Autophagy Axis in EBV-Positive Epithelial Cancers. Cancers (Basel) 2018, 10, doi:10.3390/cancers10120505.
- Gonnella, R.; Granato, M.; Farina, A.; Santarelli, R.; Faggioni, A.; Cirone, M. PKC theta and p38 MAPK activate the EBV lytic cycle through autophagy induction. Biochim. Biophys. Acta 2015, 1853, 1586–1595, doi:10.1016/j.bbamcr.2015.03.011.
- Yang, E.V.; Webster Marketon, J.I.; Chen, M.; Lo, K.W.; Kim, S.J.; Glaser, R. Glucocorticoids activate Epstein Barr virus lytic replication through the upregulation of immediate early BZLF1 gene expression. Brain Behav. Immun. 2010, 24, 1089–1096, doi:10.1016/j.bbi.2010.04.013.
- Kurokawa, M.; Ghosh, S.K.; Ramos, J.C.; Mian, A.M.; Toomey, N.L.; Cabral, L.; Whitby, D.; Barber, G.N.; Dittmer, D.P.; Harrington, W.J., Jr. Azidothymidine inhibits NF-kappaB and induces Epstein-Barr virus gene expression in Burkitt lymphoma. Blood 2005, 106, 235–240, doi:10.1182/blood-2004-09-3748.
- Liu, S.F.; Wang, H.; Li, Z.J.; Deng, X.Y.; Xiang, H.; Tao, Y.G.; Li, W.; Tang, M.; Cao, Y. Aspirin induces lytic cytotoxicity in Epstein-Barr virus-positive cells. Eur. J. Pharmacol. 2008, 589, 8–13, doi:10.1016/j.ejphar.2008.04.025.
- Ramayanti, O.; Brinkkemper, M.; Verkuijlen, S.; Ritmaleni, L.; Go, M.L.; Middeldorp, J.M. Curcuminoids as EBV Lytic Activators for Adjuvant Treatment in EBV-Positive Carcinomas. Cancers (Basel) 2018, 10, doi:10.3390/cancers10040089.
- Choi, C.K.; Ho, D.N.; Hui, K.F.; Kao, R.Y.; Chiang, A.K. Identification of Novel Small Organic Compounds with Diverse Structures for the Induction of Epstein-Barr Virus (EBV) Lytic Cycle in EBV-Positive Epithelial Malignancies. PLoS ONE 2015, 10, e0145994, doi:10.1371/journal.pone.0145994.
- Tikhmyanova, N.; Schultz, D.C.; Lee, T.; Salvino, J.M.; Lieberman, P.M. Identification of a new class of small molecules that efficiently reactivate latent Epstein-Barr Virus. ACS Chem. Biol. 2014, 9, 785–795, doi:10.1021/cb4006326.
- Stoker, S.D.; Novalic, Z.; Wildeman, M.A.; Huitema, A.D.; Verkuijlen, S.A.; Juwana, H.; Greijer, A.E.; Tan, I.B.; Middeldorp, J.M.; de Boer, J.P. Epstein-Barr virus-targeted therapy in nasopharyngeal carcinoma. J. Cancer Res. Clin. Oncol. 2015, 141, 1845–1857, doi:10.1007/s00432-015-1969-3.
- Hui, K.F.; Ho, D.N.; Tsang, C.M.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940, doi:10.1002/ijc.27439.
- Yiu, S.P.T.; Hui, K.F.; Munz, C.; Lo, K.W.; Tsao, S.W.; Kao, R.Y.T.; Yang, D.; Chiang, A.K.S. Autophagy-Dependent Reactivation of Epstein-Barr Virus Lytic Cycle and Combinatorial Effects of Autophagy-Dependent and Independent Lytic Inducers in Nasopharyngeal Carcinoma. Cancers (Basel) 2019, 11, doi:10.3390/cancers11121871.
- Hui, K.F.; Chiang, A.K. Suberoylanilide hydroxamic acid induces viral lytic cycle in Epstein-Barr virus-positive epithelial malignancies and mediates enhanced cell death. Int. J. Cancer 2010, 126, 2479–2489, doi:10.1002/ijc.24945.
- Feng, W.H.; Kenney, S.C. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expression. Cancer Res. 2006, 66, 8762–8769, doi:10.1158/0008-5472.CAN-06-1006.
- Countryman, J.K.; Gradoville, L.; Miller, G. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein-Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. J. Virol. 2008, 82, 4706–4719, doi:10.1128/JVI.00116-08.
- Gradoville, L.; Kwa, D.; El-Guindy, A.; Miller, G. Protein kinase C-independent activation of the Epstein-Barr virus lytic cycle. J. Virol. 2002, 76, 5612–5626.
- Luka, J.; Kallin, B.; Klein, G. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 1979, 94, 228–231, doi:10.1016/0042-6822(79)90455-0.
- Kim, S.J.; Kim, J.H.; Ki, C.S.; Ko, Y.H.; Kim, J.S.; Kim, W.S. Epstein-Barr virus reactivation in extranodal natural killer/T-cell lymphoma patients: A previously unrecognized serious adverse event in a pilot study with romidepsin. Ann. Oncol. 2016, 27, 508–513, doi:10.1093/annonc/mdv596.
- Tikhmyanova, N.; Paparoidamis, N.; Romero-Masters, J.; Feng, X.; Mohammed, F.S.; Reddy, P.A.N.; Kenney, S.C.; Lieberman, P.M.; Salvino, J.M. Development of a novel inducer for EBV lytic therapy. Bioorg Med. Chem. Lett. 2019, 29, 2259–2264, doi:10.1016/j.bmcl.2019.06.034.
- Moore, S.M.; Cannon, J.S.; Tanhehco, Y.C.; Hamzeh, F.M.; Ambinder, R.F. Induction of Epstein-Barr virus kinases to sensitize tumor cells to nucleoside analogues. Antimicrob. Agents Chemother. 2001, 45, 2082–2091, doi:10.1128/AAC.45.7.2082-2091.2001.
- Daigle, D.; Gradoville, L.; Tuck, D.; Schulz, V.; Wang'ondu, R.; Ye, J.; Gorres, K.; Miller, G. Valproic acid antagonizes the capacity of other histone deacetylase inhibitors to activate the Epstein-barr virus lytic cycle. J. Virol. 2011, 85, 5628–5643, doi:10.1128/JVI.02659-10.
- Le, N.T.; Richardson, D.R. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: A link between iron metabolism and proliferation. Blood 2004, 104, 2967–2975, doi:10.1182/blood-2004-05-1866.
- Iempridee, T.; Reusch, J.A.; Riching, A.; Johannsen, E.C.; Dovat, S.; Kenney, S.C.; Mertz, J.E. Epstein-Barr virus utilizes Ikaros in regulating its latent-lytic switch in B cells. J. Virol. 2014, 88, 4811–4827, doi:10.1128/JVI.03706-13.
- Robinson, A.R.; Kwek, S.S.; Kenney, S.C. The B-cell specific transcription factor, Oct-2, promotes Epstein-Barr virus latency by inhibiting the viral immediate-early protein, BZLF1. PLoS Pathog. 2012, 8, e1002516, doi:10.1371/journal.ppat.1002516.
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, doi:10.3390/cells7120278.
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, doi:10.1101/cshperspect.a008813.
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. Camb. Philos. Soc. 2019, 94, 503–516, doi:10.1111/brv.12464.
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596, doi:10.1126/science.1104904.
- Bhattacharjee, S.; Bose, P.; Patel, K.; Roy, S.G.; Gain, C.; Gowda, H.; Robertson, E.S.; Saha, A. Transcriptional and epigenetic modulation of autophagy promotes EBV oncoprotein EBNA3C induced B-cell survival. Cell Death Dis. 2018, 9, 605, doi:10.1038/s41419-018-0668-9.
- Fotheringham, J.A.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2 induces autophagy to promote abnormal acinus formation. J. Virol. 2015, 89, 6940–6944, doi:10.1128/JVI.03371-14.
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842, doi:10.1038/sj.onc.1210946.
- Pujals, A.; Favre, L.; Pioche-Durieu, C.; Robert, A.; Meurice, G.; Le Gentil, M.; Chelouah, S.; Martin-Garcia, N.; Le Cam, E.; Guettier, C., et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy 2015, 11, 2275–2287, doi:10.1080/15548627.2015.1115939.
- Hung, C.H.; Chen, L.W.; Wang, W.H.; Chang, P.J.; Chiu, Y.F.; Hung, C.C.; Lin, Y.J.; Liou, J.Y.; Tsai, W.J.; Hung, C.L., et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J. Virol. 2014, 88, 12133–12145, doi:10.1128/JVI.02033-14.
- De Leo, A.; Colavita, F.; Ciccosanti, F.; Fimia, G.M.; Lieberman, P.M.; Mattia, E. Inhibition of autophagy in EBV-positive Burkitt's lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. 2015, 6, e1876, doi:10.1038/cddis.2015.156.
- Wang, M.; Wu, W.; Zhang, Y.; Yao, G.; Gu, B. Rapamycin enhances lytic replication of Epstein-Barr virus in gastric carcinoma cells by increasing the transcriptional activities of immediate-early lytic promoters. Virus Res. 2018, 244, 173–180, doi:10.1016/j.virusres.2017.11.021.
- Adamson, A.L.; Le, B.T.; Siedenburg, B.D. Inhibition of mTORC1 inhibits lytic replication of Epstein-Barr virus in a cell-type specific manner. Virol. J. 2014, 11, 110, doi:10.1186/1743-422X-11-110.
- Pavese, J.M.; Krishna, S.N.; Bergan, R.C. Genistein inhibits human prostate cancer cell detachment, invasion, and metastasis. Am. J. Clin. Nutr. 2014, 100 Suppl. 1, 431S–436S, doi:10.3945/ajcn.113.071290.
- Prietsch, R.F.; Monte, L.G.; da Silva, F.A.; Beira, F.T.; Del Pino, F.A.; Campos, V.F.; Collares, T.; Pinto, L.S.; Spanevello, R.M.; Gamaro, G.D., et al. Genistein induces apoptosis and autophagy in human breast MCF-7 cells by modulating the expression of proapoptotic factors and oxidative stress enzymes. Mol. Cell Biochem. 2014, 390, 235–242, doi:10.1007/s11010-014-1974-x.
- Hasima, N.; Ozpolat, B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014, 5, e1509, doi:10.1038/cddis.2014.467.
- He, S.; Li, Q.; Jiang, X.; Lu, X.; Feng, F.; Qu, W.; Chen, Y.; Sun, H. Design of Small Molecule Autophagy Modulators: A Promising Druggable Strategy. J. Med. Chem 2018, 61, 4656–4687, doi:10.1021/acs.jmedchem.7b01019.
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234, doi:10.1101/gad.183434.111.
- Calame, K. Activation-dependent induction of Blimp-1. Curr. Opin. Immunol. 2008, 20, 259–264, doi:10.1016/j.coi.2008.04.010.
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761, doi:10.1161/01.ATV.0000258979.92828.bc.
- Wang, H.; Hertlein, E.; Bakkar, N.; Sun, H.; Acharyya, S.; Wang, J.; Carathers, M.; Davuluri, R.; Guttridge, D.C. NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol. Cell. Biol. 2007, 27, 4374–4387, doi:10.1128/MCB.02020-06.
- Luftig, M.; Yasui, T.; Soni, V.; Kang, M.S.; Jacobson, N.; Cahir-McFarland, E.; Seed, B.; Kieff, E. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2004, 101, 141–146, doi:10.1073/pnas.2237183100.
- Atkinson, P.G.; Coope, H.J.; Rowe, M.; Ley, S.C. Latent membrane protein 1 of Epstein-Barr virus stimulates processing of NF-kappa B2 p100 to p52. J. Biol. Chem. 2003, 278, 51134–51142, doi:10.1074/jbc.M304771200.
- Verhoeven, R.J.A.; Tong, S.; Zong, J.; Chen, Y.; Tsao, S.W.; Pan, J.; Chen, H. NF-kappaB Signaling Regulates Epstein-Barr Virus BamHI-Q-Driven EBNA1 Expression. Cancers (Basel) 2018, 10, doi:10.3390/cancers10040119.
- Valentine, R.; Dawson, C.W.; Hu, C.; Shah, K.M.; Owen, T.J.; Date, K.L.; Maia, S.P.; Shao, J.; Arrand, J.R.; Young, L.S., et al. Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-kappaB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol. Cancer 2010, 9, 1, doi:10.1186/1476-4598-9-1.
- Morrison, T.E.; Mauser, A.; Klingelhutz, A.; Kenney, S.C. Epstein-Barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor alpha-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1. J. Virol. 2004, 78, 544–549, doi:10.1128/jvi.78.1.544-549.2004.
- Gutsch, D.E.; Holley-Guthrie, E.A.; Zhang, Q.; Stein, B.; Blanar, M.A.; Baldwin, A.S.; Kenney, S.C. The bZIP transactivator of Epstein-Barr virus, BZLF1, functionally and physically interacts with the p65 subunit of NF-kappa B. Mol. Cell Biol. 1994, 14, 1939–1948, doi:10.1128/mcb.14.3.1939.
- Hsu, D.H.; de Waal Malefyt, R.; Fiorentino, D.F.; Dang, M.N.; Vieira, P.; de Vries, J.; Spits, H.; Mosmann, T.R.; Moore, K.W. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science 1990, 250, 830–832, doi:10.1126/science.2173142.
- Kenney, S.; Holley-Guthrie, E.; Mar, E.C.; Smith, M. The Epstein-Barr virus BMLF1 promoter contains an enhancer element that is responsive to the BZLF1 and BRLF1 transactivators. J. Virol. 1989, 63, 3878–3883.
- Chen, T.; Wang, Y.; Xu, Z.; Zou, X.; Wang, P.; Ou, X.; Li, Y.; Peng, T.; Chen, D.; Li, M., et al. Epstein-Barr virus tegument protein BGLF2 inhibits NF-kappaB activity by preventing p65 Ser536 phosphorylation. FASEB J. 2019, 33, 10563–10576, doi:10.1096/fj.201901196RR.
- Brown, H.J.; Song, M.J.; Deng, H.; Wu, T.T.; Cheng, G.; Sun, R. NF-kappaB inhibits gammaherpesvirus lytic replication. J. Virol. 2003, 77, 8532–8540, doi:10.1128/jvi.77.15.8532-8540.2003.
- Zou, P.; Kawada, J.; Pesnicak, L.; Cohen, J.I. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J. Virol. 2007, 81, 10029–10036, doi:10.1128/JVI.02241-06.
- Lung, H.L.; Kan, R.; Chau, W.Y.; Man, O.Y.; Mak, N.K.; Fong, C.H.; Shuen, W.H.; Tsao, S.W.; Lung, M.L. The anti-tumor function of the IKK inhibitor PS1145 and high levels of p65 and KLF4 are associated with the drug resistance in nasopharyngeal carcinoma cells. Sci. Rep. 2019, 9, 12064, doi:10.1038/s41598-019-48590-7.
- Verhoeven, R.J.; Tong, S.; Zhang, G.; Zong, J.; Chen, Y.; Jin, D.Y.; Chen, M.R.; Pan, J.; Chen, H. NF-kappaB Signaling Regulates Expression of Epstein-Barr Virus BART MicroRNAs and Long Noncoding RNAs in Nasopharyngeal Carcinoma. J. Virol. 2016, 90, 6475–6488, doi:10.1128/JVI.00613-16.
- Li, Y.; Zhang, Y.; Fu, M.; Yao, Q.; Zhuo, H.; Lu, Q.; Niu, X.; Zhang, P.; Pei, Y.; Zhang, K. Parthenolide induces apoptosis and lytic cytotoxicity in Epstein-Barr virus-positive Burkitt lymphoma. Mol. Med. Rep. 2012, 6, 477–482, doi:10.3892/mmr.2012.959.
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635, doi:10.1126/science.277.5332.1630.
- Raz, R.; Durbin, J.E.; Levy, D.E. Acute phase response factor and additional members of the interferon-stimulated gene factor 3 family integrate diverse signals from cytokines, interferons, and growth factors. J. Biol. Chem. 1994, 269, 24391–24395.
- Hino, R.; Uozaki, H.; Murakami, N.; Ushiku, T.; Shinozaki, A.; Ishikawa, S.; Morikawa, T.; Nakaya, T.; Sakatani, T.; Takada, K., et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009, 69, 2766–2774, doi:10.1158/0008-5472.CAN-08-3070.
- Chen, H.; Hutt-Fletcher, L.; Cao, L.; Hayward, S.D. A positive autoregulatory loop of LMP1 expression and STAT activation in epithelial cells latently infected with Epstein-Barr virus. J. Virol. 2003, 77, 4139–4148, doi:10.1128/jvi.77.7.4139-4148.2003.
- Zheng, H.; Li, L.L.; Hu, D.S.; Deng, X.Y.; Cao, Y. Role of Epstein-Barr virus encoded latent membrane protein 1 in the carcinogenesis of nasopharyngeal carcinoma. Cell Mol. Immunol 2007, 4, 185–196.
- Shair, K.H.; Bendt, K.M.; Edwards, R.H.; Bedford, E.C.; Nielsen, J.N.; Raab-Traub, N. EBV latent membrane protein 1 activates Akt, NFkappaB, and Stat3 in B cell lymphomas. PLoS Pathog. 2007, 3, e166, doi:10.1371/journal.ppat.0030166.
- Onozawa, E.; Shibayama, H.; Takada, H.; Imadome, K.I.; Aoki, S.; Yoshimori, M.; Shimizu, N.; Fujiwara, S.; Koyama, T.; Miura, O., et al. STAT3 is constitutively activated in chronic active Epstein-Barr virus infection and can be a therapeutic target. Oncotarget 2018, 9, 31077–31089, doi:10.18632/oncotarget.25780.
- Daigle, D.; Megyola, C.; El-Guindy, A.; Gradoville, L.; Tuck, D.; Miller, G.; Bhaduri-McIntosh, S. Upregulation of STAT3 marks Burkitt lymphoma cells refractory to Epstein-Barr virus lytic cycle induction by HDAC inhibitors. J. Virol. 2010, 84, 993–1004, doi:10.1128/JVI.01745-09.
- Hill, E.R.; Koganti, S.; Zhi, J.; Megyola, C.; Freeman, A.F.; Palendira, U.; Tangye, S.G.; Farrell, P.J.; Bhaduri-McIntosh, S. Signal transducer and activator of transcription 3 limits Epstein-Barr virus lytic activation in B lymphocytes. J. Virol. 2013, 87, 11438–11446, doi:10.1128/JVI.01762-13.
- Wu, T.; Wang, S.; Wu, J.; Lin, Z.; Sui, X.; Xu, X.; Shimizu, N.; Chen, B.; Wang, X. Icaritin induces lytic cytotoxicity in extranodal NK/T-cell lymphoma. J. Exp. Clin. Cancer Res. 2015, 34, 17, doi:10.1186/s13046-015-0133-x.
- Wang, C.; Wang, H.; Zhang, Y.; Guo, W.; Long, C.; Wang, J.; Liu, L.; Sun, X. Berberine inhibits the proliferation of human nasopharyngeal carcinoma cells via an Epstein-Barr virus nuclear antigen 1-dependent mechanism. Oncol. Rep. 2017, 37, 2109–2120, doi:10.3892/or.2017.5489.
- Lui, V.W.; Yau, D.M.; Wong, E.Y.; Ng, Y.K.; Lau, C.P.; Ho, Y.; Chan, J.P.; Hong, B.; Ho, K.; Cheung, C.S., et al. Cucurbitacin I elicits anoikis sensitization, inhibits cellular invasion and in vivo tumor formation ability of nasopharyngeal carcinoma cells. Carcinogenesis 2009, 30, 2085–2094, doi:10.1093/carcin/bgp253.
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H., et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497, doi:10.1016/j.ccr.2009.10.015.
- Greten, F.R.; Karin, M. Peering into the aftermath: JAKi rips STAT3 in cancer. Nat. Med. 2010, 16, 1085–1087, doi:10.1038/nm1010-1085.
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J., et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl Acad. Sci. USA 2007, 104, 7391–7396, doi:10.1073/pnas.0609757104.
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705, doi:10.1073/pnas.0409894102.
- Uehara, Y.; Mochizuki, M.; Matsuno, K.; Haino, T.; Asai, A. Novel high-throughput screening system for identifying STAT3-SH2 antagonists. Biochem. Biophys. Res. Commun. 2009, 380, 627–631, doi:10.1016/j.bbrc.2009.01.137.
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933, doi:10.1158/0008-5472.CAN-12-3175.
- Yang, J.; Deng, X.; Deng, L.; Gu, H.; Fan, W.; Cao, Y. Telomerase activation by Epstein-Barr virus latent membrane protein 1 is associated with c-Myc expression in human nasopharyngeal epithelial cells. J. Exp. Clin. Cancer Res. 2004, 23, 495–506.
- Terrin, L.; Dal Col, J.; Rampazzo, E.; Zancai, P.; Pedrotti, M.; Ammirabile, G.; Bergamin, S.; Rizzo, S.; Dolcetti, R.; De Rossi, A. Latent membrane protein 1 of Epstein-Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. J. Virol. 2008, 82, 10175–10187, doi:10.1128/JVI.00321-08.
- Giunco, S.; Celeghin, A.; Gianesin, K.; Dolcetti, R.; Indraccolo, S.; De Rossi, A. Cross talk between EBV and telomerase: The role of TERT and NOTCH2 in the switch of latent/lytic cycle of the virus. Cell Death Dis. 2015, 6, e1774, doi:10.1038/cddis.2015.145.
- Giunco, S.; Dolcetti, R.; Keppel, S.; Celeghin, A.; Indraccolo, S.; Dal Col, J.; Mastorci, K.; De Rossi, A. hTERT inhibition triggers Epstein-Barr virus lytic cycle and apoptosis in immortalized and transformed B cells: A basis for new therapies. Clin. Cancer Res. 2013, 19, 2036–2047, doi:10.1158/1078-0432.CCR-12-2537.
- Bryan, C.; Rice, C.; Hoffman, H.; Harkisheimer, M.; Sweeney, M.; Skordalakes, E. Structural Basis of Telomerase Inhibition by the Highly Specific BIBR1532. Structure 2015, 23, 1934–1942, doi:10.1016/j.str.2015.08.006.
- Pascolo, E.; Wenz, C.; Lingner, J.; Hauel, N.; Priepke, H.; Kauffmann, I.; Garin-Chesa, P.; Rettig, W.J.; Damm, K.; Schnapp, A. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J. Biol. Chem. 2002, 277, 15566–15572, doi:10.1074/jbc.M201266200.
- Damm, K.; Hemmann, U.; Garin-Chesa, P.; Hauel, N.; Kauffmann, I.; Priepke, H.; Niestroj, C.; Daiber, C.; Enenkel, B.; Guilliard, B., et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001, 20, 6958–6968, doi:10.1093/emboj/20.24.6958.
- El-Daly, H.; Kull, M.; Zimmermann, S.; Pantic, M.; Waller, C.F.; Martens, U.M. Selective cytotoxicity and telomere damage in leukemia cells using the telomerase inhibitor BIBR1532. Blood 2005, 105, 1742–1749, doi:10.1182/blood-2003-12-4322.
- Celeghin, A.; Giunco, S.; Freguja, R.; Zangrossi, M.; Nalio, S.; Dolcetti, R.; De Rossi, A. Short-term inhibition of TERT induces telomere length-independent cell cycle arrest and apoptotic response in EBV-immortalized and transformed B cells. Cell Death Dis. 2016, 7, e2562, doi:10.1038/cddis.2016.425.
- Seimiya, H.; Oh-hara, T.; Suzuki, T.; Naasani, I.; Shimazaki, T.; Tsuchiya, K.; Tsuruo, T. Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991. Mol. Cancer Ther. 2002, 1, 657–665.
- Serrano, D.; Bleau, A.M.; Fernandez-Garcia, I.; Fernandez-Marcelo, T.; Iniesta, P.; Ortiz-de-Solorzano, C.; Calvo, A. Inhibition of telomerase activity preferentially targets aldehyde dehydrogenase-positive cancer stem-like cells in lung cancer. Mol. Cancer 2011, 10, 96, doi:10.1186/1476-4598-10-96.
- Gurung, R.L.; Lim, S.N.; Low, G.K.; Hande, M.P. MST-312 Alters Telomere Dynamics, Gene Expression Profiles and Growth in Human Breast Cancer Cells. J. Nutrigenet. Nutrigenomics 2014, 7, 283–298, doi:10.1159/000381346.
- Morais, K.S.; Guimaraesb, A.F.R.; Ramos, D.A.R.; Silva, F.P.; de Oliveira, D.M. Long-term exposure to MST-312 leads to telomerase reverse transcriptase overexpression in MCF-7 breast cancer cells. Anticancer Drugs 2017, 28, 750–756, doi:10.1097/CAD.0000000000000508.
- Ichikawa, T.; Okuno, Y.; Sato, Y.; Goshima, F.; Yoshiyama, H.; Kanda, T.; Kimura, H.; Murata, T. Regulation of Epstein-Barr Virus Life Cycle and Cell Proliferation by Histone H3K27 Methyltransferase EZH2 in Akata Cells. mSphere 2018, 3, doi:10.1128/mSphere.00478-18.
- Ahmad, F.; Dixit, D.; Sharma, V.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis- 2016, 7, e2213, doi:10.1038/cddis.2016.117.
- Ahmad, F.; Patrick, S.; Sheikh, T.; Sharma, V.; Pathak, P.; Malgulwar, P.B.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Telomerase reverse transcriptase (TERT)-enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. J. Neurochem. 2017, 143, 671–683, doi:10.1111/jnc.14152.
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233, doi:10.1016/j.cell.2009.03.045.
- Kohlhof, H.; Hampel, F.; Hoffmann, R.; Burtscher, H.; Weidle, U.H.; Holzel, M.; Eick, D.; Zimber-Strobl, U.; Strobl, L.J. Notch1, Notch2, and Epstein-Barr virus-encoded nuclear antigen 2 signaling differentially affects proliferation and survival of Epstein-Barr virus-infected B cells. Blood 2009, 113, 5506–5515, doi:10.1182/blood-2008-11-190090.
- Anderson, L.J.; Longnecker, R. An auto-regulatory loop for EBV LMP2A involves activation of Notch. Virology 2008, 371, 257–266, doi:10.1016/j.virol.2007.10.009.
- Rowe, M.; Raithatha, S.; Shannon-Lowe, C. Counteracting effects of cellular Notch and Epstein-Barr virus EBNA2: Implications for stromal effects on virus-host interactions. J. Virol. 2014, 88, 12065–12076, doi:10.1128/JVI.01431-14.
- Gasperini, P.; Espigol-Frigole, G.; McCormick, P.J.; Salvucci, O.; Maric, D.; Uldrick, T.S.; Polizzotto, M.N.; Yarchoan, R.; Tosato, G. Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through Notch-dependent signaling. Cancer Res. 2012, 72, 1157–1169, doi:10.1158/0008-5472.CAN-11-3067.
- Li, S.; Hu, H.; He, Z.; Liang, D.; Sun, R.; Lan, K. Fine-Tuning of the Kaposi's Sarcoma-Associated Herpesvirus Life Cycle in Neighboring Cells through the RTA-JAG1-Notch Pathway. PLoS Pathog. 2016, 12, e1005900, doi:10.1371/journal.ppat.1005900.
- Yao, J.; Duan, L.; Fan, M.; Wu, X. Gamma-secretase inhibitors exerts antitumor activity via down-regulation of Notch and Nuclear factor kappa B in human tongue carcinoma cells. Oral Dis. 2007, 13, 555–563, doi:10.1111/j.1601-0825.2006.01334.x.
- Dang, C.V.; O'Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264, doi:10.1016/j.semcancer.2006.07.014.
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645, doi:10.1038/nrm1703.
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Puri, R.; Wolinsky, E.; Schineller, M., et al. MYC Controls the Epstein-Barr Virus Lytic Switch. Mol. Cell 2020, 10.1016/j.molcel.2020.03.025, doi:10.1016/j.molcel.2020.03.025.
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct. Target. Ther. 2018, 3, 5, doi:10.1038/s41392-018-0008-7.
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J., et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917, doi:10.1016/j.cell.2011.08.017.
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I., et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073, doi:10.1038/nature09504.
- Tavana, O.; Li, D.; Dai, C.; Lopez, G.; Banerjee, D.; Kon, N.; Chen, C.; Califano, A.; Yamashiro, D.J.; Sun, H., et al. HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nat. Med. 2016, 22, 1180–1186, doi:10.1038/nm.4180.
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schuttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H., et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78, doi:10.1016/j.ccr.2008.12.005.
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. U S A 2002, 99, 3830–3835, doi:10.1073/pnas.062036999.
- Keck, K.M.; Moquin, S.A.; He, A.; Fernandez, S.G.; Somberg, J.J.; Liu, S.M.; Martinez, D.M.; Miranda, J.L. Bromodomain and extraterminal inhibitors block the Epstein-Barr virus lytic cycle at two distinct steps. J. Biol. Chem. 2017, 292, 13284–13295, doi:10.1074/jbc.M116.751644.
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B., et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620, doi:10.1038/nature13393.
- Hutterer, C.; Eickhoff, J.; Milbradt, J.; Korn, K.; Zeittrager, I.; Bahsi, H.; Wagner, S.; Zischinsky, G.; Wolf, A.; Degenhart, C., et al. A novel CDK7 inhibitor of the Pyrazolotriazine class exerts broad-spectrum antiviral activity at nanomolar concentrations. Antimicrob. Agents Chemother. 2015, 59, 2062–2071, doi:10.1128/AAC.04534-14.
- Yuan, J.; Jiang, Y.Y.; Mayakonda, A.; Huang, M.; Ding, L.W.; Lin, H.; Yu, F.; Lu, Y.; Loh, T.K.S.; Chow, M., et al. Super-Enhancers Promote Transcriptional Dysregulation in Nasopharyngeal Carcinoma. Cancer Res. 2017, 77, 6614–6626, doi:10.1158/0008-5472.CAN-17-1143.
- Bark-Jones, S.J.; Webb, H.M.; West, M.J. EBV EBNA 2 stimulates CDK9-dependent transcription and RNA polymerase II phosphorylation on serine 5. Oncogene 2006, 25, 1775–1785, doi:10.1038/sj.onc.1209205.
- Sato, Y.; Watanabe, T.; Suzuki, C.; Abe, Y.; Masud, H.; Inagaki, T.; Yoshida, M.; Suzuki, T.; Goshima, F.; Adachi, J., et al. S-Like-Phase Cyclin-Dependent Kinases Stabilize the Epstein-Barr Virus BDLF4 Protein To Temporally Control Late Gene Transcription. J. Virol. 2019, 93, doi:10.1128/JVI.01707-18.
- Jeong, K.C.; Kim, K.T.; Seo, H.H.; Shin, S.P.; Ahn, K.O.; Ji, M.J.; Park, W.S.; Kim, I.H.; Lee, S.J.; Seo, H.K. Intravesical instillation of c-MYC inhibitor KSI-3716 suppresses orthotopic bladder tumor growth. J. Urol. 2014, 191, 510–518, doi:10.1016/j.juro.2013.07.019.
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K., et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497 e415, doi:10.1016/j.ccell.2019.10.001.
- Choi, S.H.; Mahankali, M.; Lee, S.J.; Hull, M.; Petrassi, H.M.; Chatterjee, A.K.; Schultz, P.G.; Jones, K.A.; Shen, W. Targeted Disruption of Myc-Max Oncoprotein Complex by a Small Molecule. ACS Chem. Biol. 2017, 12, 2715–2719, doi:10.1021/acschembio.7b00799.
- Muller, I.; Larsson, K.; Frenzel, A.; Oliynyk, G.; Zirath, H.; Prochownik, E.V.; Westwood, N.J.; Henriksson, M.A. Targeting of the MYCN protein with small molecule c-MYC inhibitors. PLoS ONE 2014, 9, e97285, doi:10.1371/journal.pone.0097285.
- Castell, A.; Yan, Q.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J., et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 10064, doi:10.1038/s41598-018-28107-4.
- Yu, C.; Niu, X.; Jin, F.; Liu, Z.; Jin, C.; Lai, L. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep. 2016, 6, 22298, doi:10.1038/srep22298.
- Yao, R.; Sun, X.; Xie, Y.; Sun, X.; Yao, Y.; Li, H.; Li, Z.; Gao, J.; Xu, K. Identification of a novel c-Myc inhibitor with antitumor effects on multiple myeloma cells. Biosci. Rep. 2018, 38, doi:10.1042/BSR20181027.
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E., et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119, doi:10.1016/j.ejmech.2018.09.023.
- Jung, K.Y.; Wang, H.; Teriete, P.; Yap, J.L.; Chen, L.; Lanning, M.E.; Hu, A.; Lambert, L.J.; Holien, T.; Sundan, A., et al. Perturbation of the c-Myc-Max protein-protein interaction via synthetic alpha-helix mimetics. J. Med. Chem. 2015, 58, 3002–3024, doi:10.1021/jm501440q.
- Carvalho, A.L.; Trincao, J.; Romao, M.J. X-ray crystallography in drug discovery. Methods Mol. Biol. 2009, 572, 31–56, doi:10.1007/978-1-60761-244-5_3.
- Sugiki, T.; Furuita, K.; Fujiwara, T.; Kojima, C. Current NMR Techniques for Structure-Based Drug Discovery. Molecules 2018, 23, doi:10.3390/molecules23010148.
- Renaud, J.P.; Chari, A.; Ciferri, C.; Liu, W.T.; Remigy, H.W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471–492, doi:10.1038/nrd.2018.77.
- Jiang, L.; Lung, H.L.; Huang, T.; Lan, R.; Zha, S.; Chan, L.S.; Thor, W.; Tsoi, T.H.; Chau, H.F.; Borestrom, C., et al. Reactivation of Epstein-Barr virus by a dual-responsive fluorescent EBNA1-targeting agent with Zn(2+)-chelating function. Proc. Natl. Acad. Sci. USA 2019, 10.1073/pnas.1915372116, doi:10.1073/pnas.1915372116.
- Jiang, L.; Xie, C.; Lung, H.L.; Lo, K.W.; Law, G.L.; Mak, N.K.; Wong, K.L. EBNA1-targeted inhibitors: Novel approaches for the treatment of Epstein-Barr virus-associated cancers. Theranostics 2018, 8, 5307–5319, doi:10.7150/thno.26823.
- Zha, S.; Fung, Y.H.; Chau, H.F.; Ma, P.; Lin, J.; Wang, J.; Chan, L.S.; Zhu, G.; Lung, H.L.; Wong, K.L. Responsive upconversion nanoprobe for monitoring and inhibition of EBV-associated cancers via targeting EBNA1. Nanoscale 2018, 10, 15632–15640, doi:10.1039/c8nr05015e.
- Jiang, L.; Lui, Y.L.; Li, H.; Chan, C.F.; Lan, R.; Chan, W.L.; Lau, T.C.; Tsao, G.S.; Mak, N.K.; Wong, K.L. EBNA1-specific luminescent small molecules for the imaging and inhibition of latent EBV-infected tumor cells. Chem. Commun. (Camb) 2014, 50, 6517–6519, doi:10.1039/c4cc01589d.
- Latchman, D.S. The Oct-2 transcription factor. Int. J. Biochem. Cell Biol. 1996, 28, 1081–1083, doi:10.1016/1357-2725(96)00050-7.
- Medvedovic, J.; Ebert, A.; Tagoh, H.; Busslinger, M. Pax5: A master regulator of B cell development and leukemogenesis. Adv. Immunol. 2011, 111, 179–206, doi:10.1016/B978-0-12-385991-4.00005-2.
- Raver, R.M.; Panfil, A.R.; Hagemeier, S.R.; Kenney, S.C. The B-cell-specific transcription factor and master regulator Pax5 promotes Epstein-Barr virus latency by negatively regulating the viral immediate early protein BZLF1. J. Virol. 2013, 87, 8053–8063, doi:10.1128/JVI.00546-13.
- Chia, M.C.; Leung, A.; Krushel, T.; Alajez, N.M.; Lo, K.W.; Busson, P.; Klamut, H.J.; Bastianutto, C.; Liu, F.F. Nuclear factor-Y and Epstein Barr virus in nasopharyngeal cancer. Clin. Cancer Res. 2008, 14, 984–994, doi:10.1158/1078-0432.CCR-07-0828.
- Ma, Y.; Walsh, M.J.; Bernhardt, K.; Ashbaugh, C.W.; Trudeau, S.J.; Ashbaugh, I.Y.; Jiang, S.; Jiang, C.; Zhao, B.; Root, D.E., et al. CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 2017, 21, 580–591 e587, doi:10.1016/j.chom.2017.04.005.
- Zhou, T.; Schmidt, J.; Kim, Y.; MacLeod, A.R. Therapeutic Targeting of Interferon Regulatory Factor 4 with Next Generation Antisense Oligonucleotides Produces Robust In Vivo Antitumor Activity in Preclinical Models of Multiple Myeloma. Blood 2017, 130, 3078–3078, doi:10.1182/blood.V130.Suppl_1.3078.3078.
- Lightbody, E.D.; Reidy, M.; Agius, M.P.; El-Behaedi, S.; Sklavenitis-Pistofidis, R.; Manier, S.; Ghobrial, I.M. A High-Throughput Drug Screen Reveals a Novel Compound Class That Significantly Depletes IRF4 Expression in Multiple Myeloma. Blood 2019, 134, 5545–5545, doi:10.1182/blood-2019-130910.
- Chang, Y.H.; Lee, C.P.; Su, M.T.; Wang, J.T.; Chen, J.Y.; Lin, S.F.; Tsai, C.H.; Hsieh, M.J.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase retards cellular S-phase progression and induces chromosomal abnormality. PLoS ONE 2012, 7, e39217, doi:10.1371/journal.pone.0039217.
- Chiu, S.H.; Wu, C.C.; Fang, C.Y.; Yu, S.L.; Hsu, H.Y.; Chow, Y.H.; Chen, J.Y. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 2014, 5, 8583–8601, doi:10.18632/oncotarget.2323.
- Wu, C.C.; Liu, M.T.; Chang, Y.T.; Fang, C.Y.; Chou, S.P.; Liao, H.W.; Kuo, K.L.; Hsu, S.L.; Chen, Y.R.; Wang, P.W., et al. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949, doi:10.1093/nar/gkp1169.
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J., et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257, doi:10.1038/ncomms14257.
- Whitehurst, C.B.; Vaziri, C.; Shackelford, J.; Pagano, J.S. Epstein-Barr virus BPLF1 deubiquitinates PCNA and attenuates polymerase eta recruitment to DNA damage sites. J. Virol. 2012, 86, 8097–8106, doi:10.1128/JVI.00588-12.


