Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Megan Ann Opichka | + 3475 word(s) | 3475 | 2021-11-08 03:02:53 | | | |
| 2 | Peter Tang | Meta information modification | 3475 | 2021-11-17 03:14:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Opichka, M. Vascular Dysfunction in Preeclampsia. Encyclopedia. Available online: https://encyclopedia.pub/entry/16059 (accessed on 24 June 2026).
Opichka M. Vascular Dysfunction in Preeclampsia. Encyclopedia. Available at: https://encyclopedia.pub/entry/16059. Accessed June 24, 2026.
Opichka, Megan. "Vascular Dysfunction in Preeclampsia" Encyclopedia, https://encyclopedia.pub/entry/16059 (accessed June 24, 2026).
Opichka, M. (2021, November 16). Vascular Dysfunction in Preeclampsia. In Encyclopedia. https://encyclopedia.pub/entry/16059
Opichka, Megan. "Vascular Dysfunction in Preeclampsia." Encyclopedia. Web. 16 November, 2021.
Copy Citation
Preeclampsia is a life-threatening pregnancy-associated cardiovascular disorder characterized by hypertension and proteinuria at 20 weeks of gestation. Potential abnormalities include impaired placentation, incomplete spiral artery remodeling, and endothelial damage, which are further propagated by immune factors, mitochondrial stress, and an imbalance of pro- and antiangiogenic substances. Newfound evidence indicates that vasopressin is an early mediator and biomarker of the disorder, and promising future therapeutic avenues include mitigating mitochondrial dysfunction, excess oxidative stress, and the resulting inflammatory state.
preeclampsia
pregnancy
gestation
hypertension
vessel
blood pressure
placenta
trophoblast
1. Introduction
Preeclampsia is a pregnancy-related hypertensive disorder and a major cause of maternal and perinatal morbidity and mortality [1][2]. Despite its prevalence, well-cataloged risk factors, and clinical characteristics, the exact pathophysiology of this disorder remains unknown [2]. This deficit in knowledge has hampered the development of targeted therapies and limited treatment options for healthcare providers.
Clinically, preeclampsia is associated with a number of complications for both mother and fetus [3]. It is thought to lie on a spectrum of hypertensive diseases in pregnancy, with gestational hypertension at the mildest end of the spectrum, followed by preeclampsia, chronic hypertension with superimposed preeclampsia, hemolysis, elevated liver enzymes, low platelet count (HELLP) syndrome, and eclampsia at the most extreme end [1] (Figure 1). As preeclampsia is a true systemic disease, it may manifest in a number of different ways. Classically, it has been defined as maternal hypertension and renal dysfunction, specifically characterized by proteinuria [1]. However, more recent guidelines have noted thrombocytopenia, impaired liver function, pulmonary edema, and cerebral/visual symptoms as diagnostic features [1][2][4]. Additional maternal complications include seizures (eclampsia), cerebral hemorrhage, disseminated intravascular coagulation, and hepatic rupture [4]. Obstetric complications associated with preeclampsia consist of uteroplacental insufficiency, placental abruption, prematurity, and increased risk of cesarean delivery [2]. Additional fetal complications include intrapartum fetal distress, intrauterine growth restriction, oligohydramnios, and in severe cases, stillbirth [3]. Epidemiologic studies have linked approximately 15–20% of all fetal growth restriction and small for gestational age infants to preeclampsia, while 20% of all preterm births are associated with the disease [5][6]. Beyond the obstetric and neonatal consequences, preeclampsia confers long-term risk of complications, including cerebrovascular accident and hypertension [7][8].

Figure 1. Spectrum of hypertensive disorders during pregnancy and their prevalence [9][10][11]. Gestational hypertension is defined by new-onset elevations in blood pressure (<140/90 mmHg) after 20 weeks of gestation, whereas preeclampsia is also accompanied by proteinuria and end-organ dysfunction. Chronic hypertension is present prior to 20 weeks of gestation or continues >12 weeks into the postnatal period and can occur in concert with preeclampsia. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome is classified as a subset of preeclampsia, and eclampsia is a complication of preeclampsia characterized by the addition of seizures.
An awareness of the syndrome dates back to Hippocrates in ~400 BC, yet preeclampsia remains a significant obstetric concern [12]. According to the World Health Organization, 16% of maternal deaths are attributable to preeclampsia and related gestational hypertensive disorders [13]. Furthermore, multiple studies have displayed an alarming increase in the incidence of gestational hypertension and preeclampsia over the past 3 decades [6][14][15][16]. Combining data from the Agency for Healthcare Research and Quality, the Center for Delivery, Organization, and Markets, Healthcare Cost and Utilization Project, and the National Inpatient Sample, the rate of overall preeclampsia and eclampsia increased by 21% from 2004 to 2014, with the incidence of severe preeclampsia rising by 50% during this period [11]. This imposes a severe clinical and economic burden, as the annual total cost associated with maternal and neonatal consequences of preeclampsia 12 months after delivery was USD 2.18 billion in the United States in 2012 [17].
As clinicians attempt to better manage this rising tide, a number of risk factors have been identified that reflect the complex nature of preeclampsia [15][18]. These include conditions such as chronic hypertension and other classical cardiovascular risk factors, along with chronic renal disease, antiphospholipid syndrome, collagen vascular diseases (e.g., lupus), and preexisting diabetes [18]. Additionally, factors such as nulliparity, a previous diagnosis of preeclampsia, abnormal placentation, multiple gestation, and maternal age at either end of the spectrum (<20 years or >35 years) also increase susceptibility [15]. The rate and severity of preeclampsia are higher among African Americans [19][20], and this likely reflects healthcare inequities as well as a higher incidence of determinants, including chronic hypertension, obesity, and type 2 diabetes, which are underdiagnosed in the African American community [21]. Finally, there appears to be a genetic component to preeclampsia, as a family history of preeclampsia, hypertension, and type II diabetes (either maternal or paternal) also portends higher risk [22][23].
Nonetheless, despite health disparities and the significant clinical impact of preeclampsia, the only “cure” is delivery, and even after childbirth, there remains an elevated risk of cardiovascular and metabolic disease later in life for these mothers and their children [24][25][26][27][28]. Thus, efforts to facilitate early detection, a better understanding of gestational mechanisms, and enhanced treatment modalities are imperative for improved management and health outcomes in patients with this complex condition. As the field advances, there is an increasing awareness that multiple subtypes of preeclampsia exist, and these subtypes may vary in their underlying cause, placental transcriptomic landscape, and disease severity [29][30][31][32]. Proposed classifications include early vs. late onset, in which early is more commonly associated with malplacentation, poor uterine perfusion, and fetal growth restriction. Alternatively, late-onset preeclampsia may be a consequence of placental overgrowth (resulting in compression of the chorionic villi), stress, or senescence towards the end of pregnancy [29][30][31][32].
2. Pathophysiology of Preeclampsia
2.1. Improper Decidualization and Placentation
It is widely accepted that placental development is disrupted in some pregnancies affected by preeclampsia, leading to cellular, molecular, immunological, and vascular changes [33][34][35], and the role of insufficient decidualization has also received increasing attention [36][37][38][39]. Early-onset preeclampsia is classically thought to be mediated by abnormal placentation and shallow trophoblast invasion within the uterus, thereby resulting in incomplete spiral artery remodeling [33][34][35]. This may lead to placental hypoxia, an aberrant angiogenic state, endothelial dysfunction, further decrements in placental formation, trophoblast stress, and ultimately the maternal presence of preeclampsia [2][35][40]. While much of the etiology remains unknown, research suggests that failed decidual differentiation prior to pregnancy can contribute to impaired trophoblast invasion and its sequela [36][37][38][39].
However, this is a complex syndrome, and the exact order of events in the pathogenesis is unclear. Predicting preeclampsia is imprecise, and determining whether physiological alterations cause preeclampsia or are a secondary result is quite challenging.
3. Circulating and Placenta-Derived Vascular Substances Associated with Preeclampsia (VEGF, PlGF, sFLT-1, ENG, and sENG)
Preeclampsia is characterized by an imbalance of pro- and antiangiogenic factors, which directly impacts endothelial function [41][42][43][44]. VEGFA stimulates angiogenesis, vascular permeability, and cell migration by binding to its tyrosine kinase receptors VEGFR1 and VEGFR2 [45][46]. VEGFA binding to VEGFR2 elicits stronger signaling than VEGFR1 [47] via activation of the phospholipase C gamma (PLCy)/protein kinase C (PKC)/MAPK pathway involved in endothelial cell proliferation [45]. During placental villous development, VEGFA is present in trophoblasts and perivascular cells to support de novo vascular development (i.e., vasculogenesis), as well as vessel expansion via endothelial sprouting (i.e., angiogenesis) [48]. In pregnancy, VEGF induces a more robust activation of endothelial nitric oxide synthase (eNOS), with NO production primarily occurring through VEGFR2-induced PI3K/AKT signaling [49][50]. PlGF, its proangiogenic counterpart, binds to VEGFR1 to increase the likelihood of VEGF binding to VEGFR-2 [51]. PlGF’s interaction with VEGFR1 also promotes other critical events, such as transphosphorylation of VEGFR2, augmenting its downstream signaling cascade [51]. Similar to VEGF, the actions of PlGF facilitate the growth and migration of endothelial and trophoblast cells [51][52][53]. In healthy pregnancies, PlGF increases until 32 weeks and then subsequently declines [54][55]. However, in preeclampsia, there is a marked reduction in venous levels as early as 13–16 weeks, occurring before the onset of other clinical symptoms [52][56]. Not only does this have adverse cardiovascular consequences during pregnancy, but these vascular pathologies and unfavorable cardiac remodeling can persist for years beyond pregnancy [57]. This suggests that though overt symptoms are often resolved after delivery, preeclamptic mothers are at risk for cardiovascular disease years or decades later.
While the VEGFR is membrane bound, a truncated, soluble version known as soluble fms-like tyrosine kinase 1 (sFLT-1) sequesters ligands that bind to the VEGFR, specifically VEGF and PlGF [58]. sFLT-1 is similar to VEGFR1 but lacks the membrane-spanning domain [59]. sFLT-1 plays a role in the pathogenesis of preeclampsia and can be detected in the serum and placenta before other clinical manifestations, including proteinuria and hypertension [43][60]. The sFLT-1/PlGF ratio from 24 weeks to 36 weeks 6 days has been deemed a useful tool for predicting the absence of preeclampsia within 1 week of measurement, with a negative predictive value of 99.3% at a cutoff of 38 [61]. However, the positive predictive value for the diagnosis of preeclampsia within the next 4 weeks at this threshold was only 36.7% [61]. Thus, while this test is informative, earlier detection methods and tools with greater positive predictive value would be more powerful for improving the care of women prior to preeclampsia.
Adding to the complexity of this disorder, it has been discovered that a unique isoform of sFLT-1 predominates after the first trimester in both healthy and preeclamptic pregnancies [59][62]. At term, preeclamptic placentas express much higher sFLT1 mRNA, specifically in syncytial knots, which are a source of circulating sFLT [62]. Furthermore, hypoxic conditions stimulate mRNA expression of the novel sFLT1 variant and sFLT-1 peptide release in cultured primary cytotrophoblast and syncytiotrophoblast cells [59][63], but these effects are cell-specific, such that low oxygen tension does not increase the release of sFLT-1 protein in the culture medium of HUVECS or villous fibroblasts [63]. In cytotrophoblasts, no changes in free VEGF protein were detected under conditions of reduced oxygen tension despite an increase in total VEGF protein production, suggesting sequestration by sFLT-1 [63]. This imbalance leads to elevated oxidative stress, evident by increased lipid peroxidase relative to superoxide dismutase [64], a key antioxidant for scavenging oxygen free radicals [65].
Endoglin (ENG) is yet another hypoxia-induced protein implicated in preeclampsia [66][67][68]. This protein is located on endothelial cells, syncytiotrophoblasts, and columnar cytotrophoblasts prior to uterine invasion and exists in either a proangiogenic membrane-bound form or an antiangiogenic soluble form [66][67][69]. ENG serves as a receptor for the cytokines transforming growth factor-beta 1 and 3 (TGF-β1 and TGF-β3), which perform functions related to cell proliferation and apoptosis [67][70][71]. There are contradictory findings regarding the role of TGF-β in preeclampsia, which may be a product of gestational-age-related variations. Ayatollahi et al. reported no differences in serum TGF-β1 between healthy and preeclamptic pregnancies at 36–40 weeks and suggested that the anti-inflammatory properties of TGF-β may be instrumental for fetal-allograft survival [72]. Alam et al. found elevated TGF-β1 levels in preeclamptic patients from 30 to 33 weeks with a slight drop after 33 weeks [70], which may explain the lack of difference in the aforementioned study. One proposed explanation for the increase in TGF-β1 involves the shedding of necrotic placental trophoblast cells into the circulation, which are thought to undergo apoptosis in healthy pregnancies. When necrotic trophoblasts are phagocytosed by endothelial cells, the endothelial cells secrete TGF-β1, leading to IL-6 release. This causes placental soluble endoglin (sENG) secretion, sENG occupancy of ENG receptors, and impaired NO synthesis. Because TGF-β cannot bind to its receptor, its circulatory presence is increased, explaining the rise in TGF-β in preeclamptic women [70].
Both membrane and sENG are elevated in preeclampsia and have been considered as a diagnostic tool for detecting the syndrome [68][73]. Excess sENG has detrimental effects on endothelial tube development. In animal models, injection of both sENG and sFLT-1 results in a severe preeclamptic phenotype, accompanied by liver defects, fetal weight restriction, and neurological impairments [74]. More favorably, transmembrane ENG has been associated with NO-mediated vasodilation via eNOS expression [75][76] as well as trophoblast differentiation prior to invasion [71][75] (Figure 2). However, fluctuations in membrane ENG concentration and localization throughout pregnancy may be necessary for the maintenance of placental function because ENG knockdown has been shown to support extravillous trophoblast invasion [77].
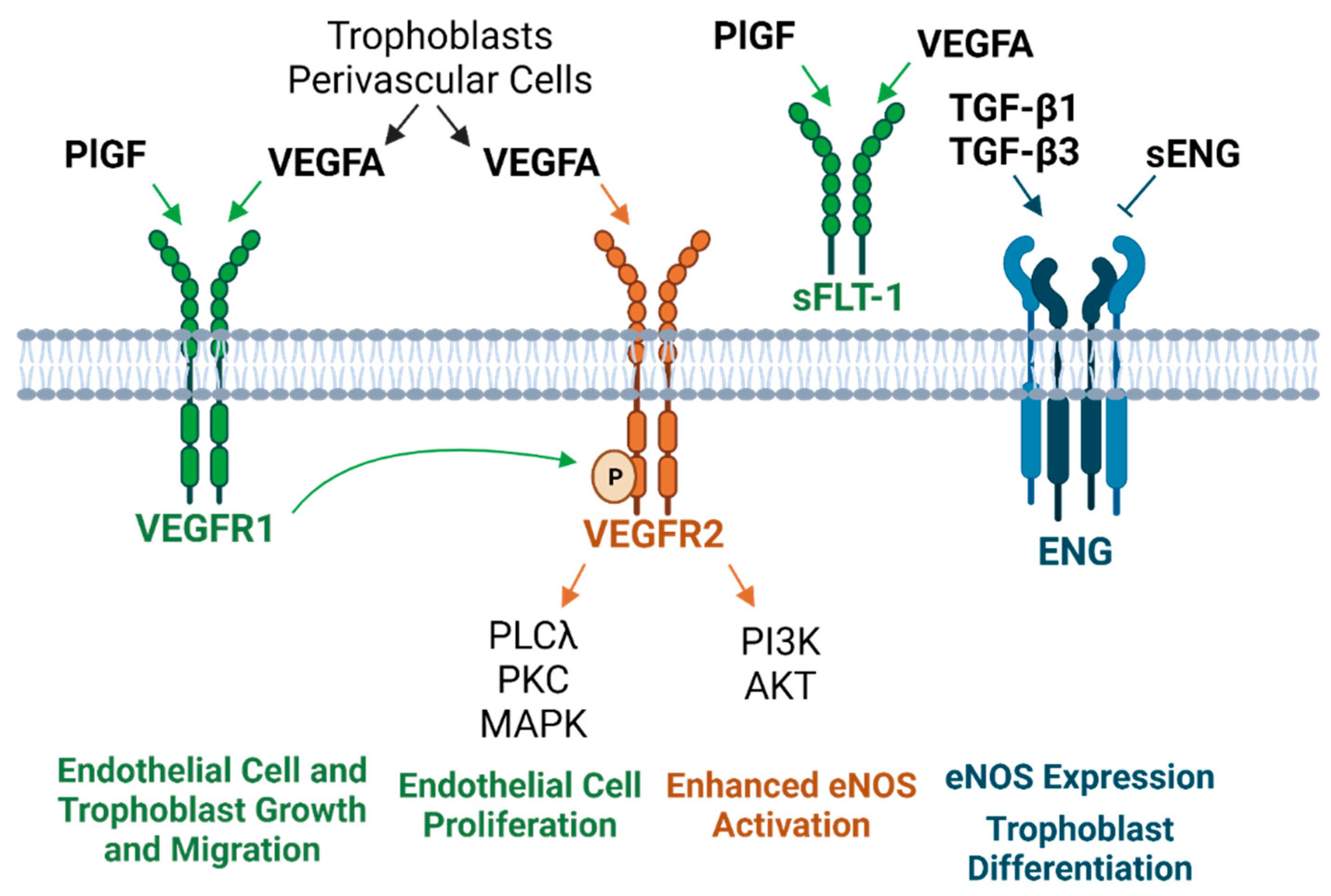
Figure 2. Circulating and placenta-derived vascular substances associated with preeclampsia and their downstream cellular effects [66][45][46][48][49][50][51][58][59][67][68][70][71][75][76][78]. PlGF, VEGFA, and ENG are considered “proangiogenic” factors, whereas sFLT-1 and ENG are “antiangiogenic.” sFLT-1 sequesters PlGF and VEGFA, and sENG blocks TGF-β binding to the ENG receptor (PlGF, placental growth factor; VEGFA, vascular endothelial growth factor A; sFLT-1, soluble fms-like tyrosine kinase 1; ENG, endoglin; sENG, soluble endoglin; VEGFR1 and VEGFR2, vascular endothelial growth factor receptor 1 and 2; TGF-β, transforming growth factor-beta; PLCλ, phospholipase C gamma; PKC, protein kinase C; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; eNOS, endothelial nitric oxide synthase).
Several hypotheses exist regarding the mechanism of elevated sENG in preeclampsia, many of which are thought to independently contribute to the disorder [66][75]. Proposed notions include insufficient endogenous oxidative protective factors, NFκB activation, the MAPK stress response, MMP-regulated cleavage from endothelial plasma membranes, and angiotensin II type 1 receptor autoantibodies (AT1-AA) [66][75]. Experimental AT1A receptor activation elicits many preeclamptic-like phenotypes, including cell proliferation, vasoconstriction, renal fluid reabsorption, and vascular smooth muscle cell (VSMC) hypertrophy [79]. Mechanistically, rat models suggest that reduced uterine perfusion pressure stimulates increases in TNFα, which is coupled to AT1AA production [80][81]. Further, AT1AA infusion induced elevations in sENG as well as endothelin-1, and sFLT-1 [82][83].
While there are no reliable preventive or treatment therapeutics for preeclampsia [84][85][86], numerous studies support the notion that proangiogenic factors may be a viable future target against the high sFLT-1 levels found in preeclampsia [43][87][88][89]. Administration of VEGF and PlGF reestablished endothelial tube formation in HUVECs after it was impeded by preeclamptic serum [43]. In vitro, VEGF and PlGF produce dose-dependent arteriole vasodilation, and this response is blocked by sFLT1 [43]. Along with compromised vasomotor function, sFLT-1 increases placental and vascular superoxide production, and vasodilatory impairment can be reversed by free-radical scavenging [90]. In vivo, animal models indicate that PlGF infusion eliminates hypertension caused by both sFLT-1 and reduced uteroplacental perfusion [87][88]. Removal of sFLT-1 from women prolonged pregnancy [89]. Therefore, reversing the ratio of circulating vascular substances in preeclampsia has the potential to restore the dysregulated homeostatic state of preeclamptic women or prevent the progression of this syndrome in those at high risk.
4. Endothelial Damage in Preeclampsia
The endothelium, or inner layer of blood vessels, serves a wide array of functions that encompass but are not constrained to hemostasis, fibrinolysis, regulation of vascular tone, mediation of inflammatory cascades, and permeability [91][92][93][94]. Endothelial dysfunction, specifically in the form of barrier disruption and impaired vasodilatory capacity, is prevalent in preeclampsia and implicated in many stages of the disease [95][96][97][98][99]. Hemodynamic shifts accompanying compromised endothelial junction integrity, specifically those related to vasopressin, may precede early malfunctions in placental development [100], or in late-onset preeclampsia, endothelial damage may render a mother unable to buffer trophoblast-derived stress signals that accumulate throughout pregnancy [29][32]. The vascular defects of late-stage preeclampsia appear to be targeted to the endothelium in certain vascular beds, which is evident by the incubation of uterine myometrial resistance vessels with preeclamptic plasma [97]. Preeclamptic plasma restricted endothelium-dependent relaxation, but the effect only occurred with an intact endothelium, demonstrating that the vascular smooth muscle was unimpaired [97]. Similarly, placental vessels obtained from preeclamptic pregnancies show attenuated endothelial function with unaltered smooth muscle-mediated dilation [98]. These endothelial-specific findings have also been confirmed in vivo, as endothelium-dependent flow-mediated dilation was impaired in women with a history of preeclampsia compared to those without [99]. Together, the imbalance between constriction and relaxation and hemodynamic modifications that alter body fluid homeostasis are prominent features of preeclampsia [101].
5. Preeclampsia-Associated Platelet Alterations
Trophoblast stress is a common feature of preeclampsia and results in a compensatory surge of inflammatory mediators [29][102][103][104][105]. Of these, prostacyclin is a product of arachidonic acid metabolism [105][106]. Its role as a vasodilatory factor opposes the action of thromboxane, which elicits vascular smooth muscle constriction and platelet aggregation [107] (Figure 3).
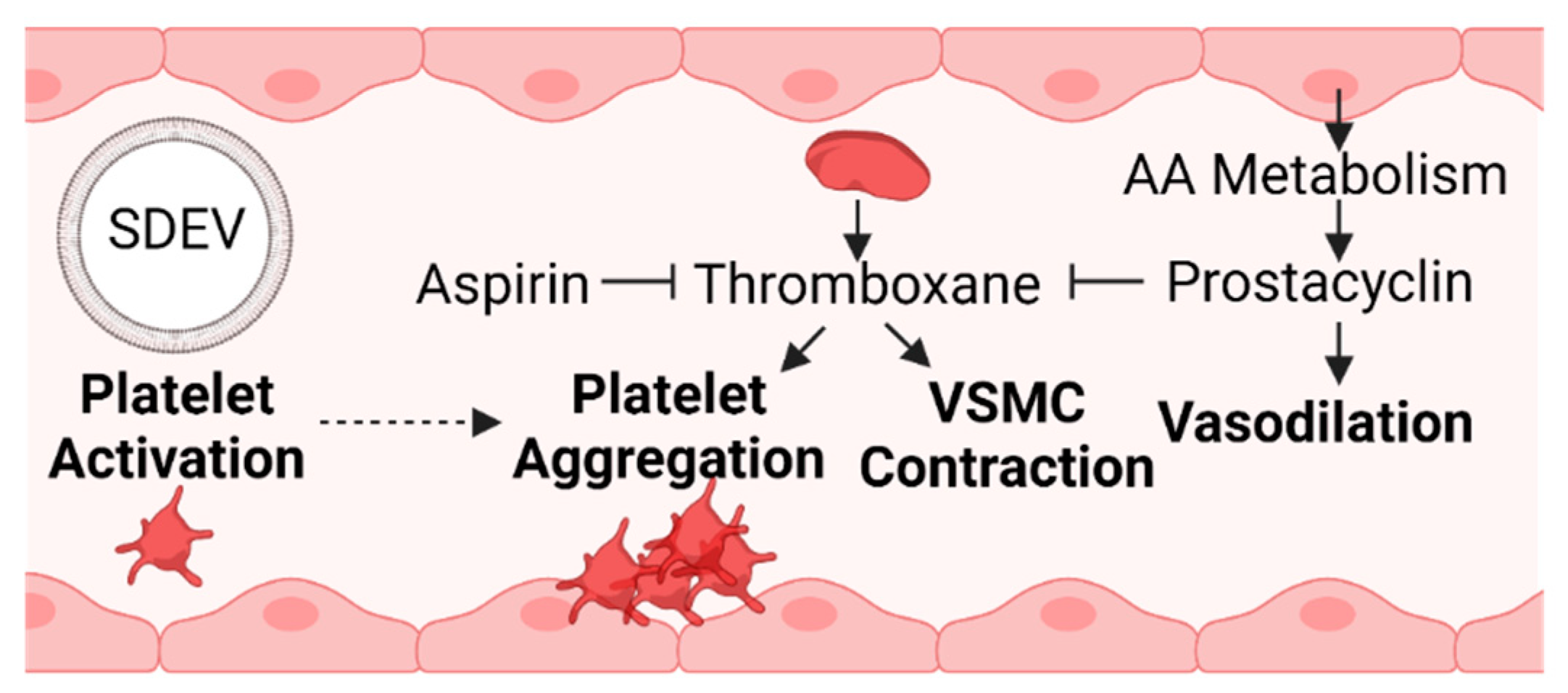
Figure 3. An imbalance in coagulation factors promotes a more prothrombotic environment during preeclampsia [107][108][109]. Prostacyclin is a vasodilatory product of arachidonic acid metabolism derived from endothelial cells [106]. Prostacyclin and aspirin oppose the actions of thromboxane, and the ratio of prostacyclin to thromboxane decreases in preeclampsia [107][110]. The accumulation of placental stress can lead to the release of SDEVs into maternal circulation. SDEVs can promote platelet activation, a precursory step to platelet aggregation and the formation of blood clots [29][111] (SDEV, syncytiotrophoblast-derived extracellular vesicle; VSMC, vascular smooth muscle cell; AA, arachidonic acid).
These two vasoactive substances are produced by the endothelium, platelets, and reproductive tissues, and the balance between them is disrupted in preeclampsia, with the level of placental and plasma thromboxane exceeding that of prostacyclin [107][108][109]. The pharmacological effects of aspirin diminish platelet thromboxane production, altering the prostaglandin thromboxane ratio [108]. A meta-analysis revealed that starting low-dose aspirin early in pregnancy as a preventative measure for preeclampsia has moderately favorable results [112]. The American College of Obstetricians and Gynecologists, the Society for Maternal-Fetal Medicine, and the U.S. Preventative Services Task Force now recommend low-dose aspirin for women with a high risk of preeclampsia [113].
Platelet activation, aggregation, and blood coagulation (clotting) are interrelated processes [114]. Briefly, platelets have adhesive properties and, upon binding to an injured endothelium, release substances such as thromboxane to promote aggregation. Platelet aggregation encourages the formation of a platelet plug and thrombin-mediated generation of a cross-linked fibrin clot [114]. A recent systemic review and meta-analysis suggests that preeclamptic patients have higher mean platelet volume (indicating platelet activation) and a higher likelihood of adhesion and aggregation [115]. In this paper, Jakobsen et al. reported inconsistent findings regarding aggregation, with more studies suggesting no difference or decreased aggregation, but these particular studies did not assess adhesion [115]. One study that did assess platelet adhesion reported decreased immunohistochemical expression of platelet endothelial cell adhesion molecule-1 and increased intercellular adhesion molecule-1 in the human placenta of preeclamptic individuals, which has been proposed to play a role in trophoblast invasion and vascular dysfunction [115][116]. Converging the idea that syncytiotrophoblast stress is a final common factor that leads to the maternal elements of preeclampsia [29] with the importance of platelet function in this syndrome, syncytiotrophoblast-derived extracellular vesicles (SDEVs) have been shown to activate platelets ex vivo [111]. SDEVs obtained from preeclamptic placentas evoke greater platelet activation than those from normal pregnancies, but platelet aggregation is prevented by aspirin treatment [111].
During pregnancy, there is a natural decline in platelet count throughout gestation [117], part of which may be attributed to sequestration of blood cells in the intervillous space [118], increases in plasma volume [119], and heightened aggregation from thromboxane A2 [120]. Thrombocytopenia beyond normal pregnancy-induced platelet decreases is commonly seen in preeclampsia and may specifically be accompanied by reduced platelet numbers [120][121][122] and activated coagulation [121][123]. Together, these hemostatic effects contribute to bleeding and microthrombi risk in preeclamptic mothers [120][124].
6. Oxidative Stress, Mitochondrial DNA Damage, and TLR9 Activation
The generation of reactive oxygen species via placental hypoxia, immune activation, and other cellular insults has numerous consequences, including mitochondrial DNA (mtDNA) damage [125][126][127][128]. The DNA repair capabilities of the mitochondria are less extensive than those for nuclear DNA, which renders cells with mtDNA mutations more susceptible to death by apoptosis or necrosis [129][130]. This causes the release of DNA into the maternal circulation, which is considered a damage-associated molecular pattern (DAMP), recognized by pattern recognition receptors such as TLR9 [131]. TLR9 is a pro-inflammatory innate immune component activated by hypomethylated CpG dinucleotides, which are prevalent in mtDNA and bacteria [132][133]. Although surface receptors are also present, the recognition of DNA by TLR9 primarily occurs in endolysosomes because the acidic environment allows TLR9 to more easily bind to negative DNA [132]. Thus, DNA enters the cell through endocytosis [134] and, upon TLR9 binding, causes a downstream proinflammatory cascade of events, including signaling for IFNs, NFκB, and AP-1 [135].
Supporting these notions, there is an increased abundance of serum mtDNA in preeclamptic plasma [136], and TLR9 activity is elevated upon the presentation of preeclamptic symptoms [128] (Figure 4). Recent findings by He et al. link the TLR9 inflammatory response to other aspects of preeclampsia, including angiogenesis and trophoblast function [135]. In this study, human placental VEGFA was decreased, but TLR9 and sFLT-1 were increased in preeclamptic samples [135]. Applying these findings to a mouse model, a TLR9 agonist induced the traditional hallmarks of preeclampsia and also reproduced the downregulated VEGFA and elevated sFLT-1 observed in human tissue [135]. siRNA knockdown of TLR9 in human trophoblast cells facilitated migration and invasion, which highlights the importance of TLR9 in early phases of placentation as well [135]. The dendritic cells of preeclamptic women appear to be hyperresponsive to immune-evoking substances, suggesting a potential source for this excess TLR9 engagement. In preeclampsia, dendritic cell TLR9 expression levels were higher, and upon stimulation, these receptors evoked more proinflammatory cytokines compared to healthy pregnant controls [137]. These data reveal an interconnected relationship among inflammation, oxidative stress, TLR9 activation, the release of antiangiogenic factors, and trophoblast dysfunction [135][125][126][129][130][131] and reiterate the complex interplay between many molecular mediators in women with preeclampsia. Understanding each of these components and how they interact is essential to mitigating the disorder but has proven difficult considering its heterogeneity and the lack of a single, well-understood initiating mechanism [29][31].
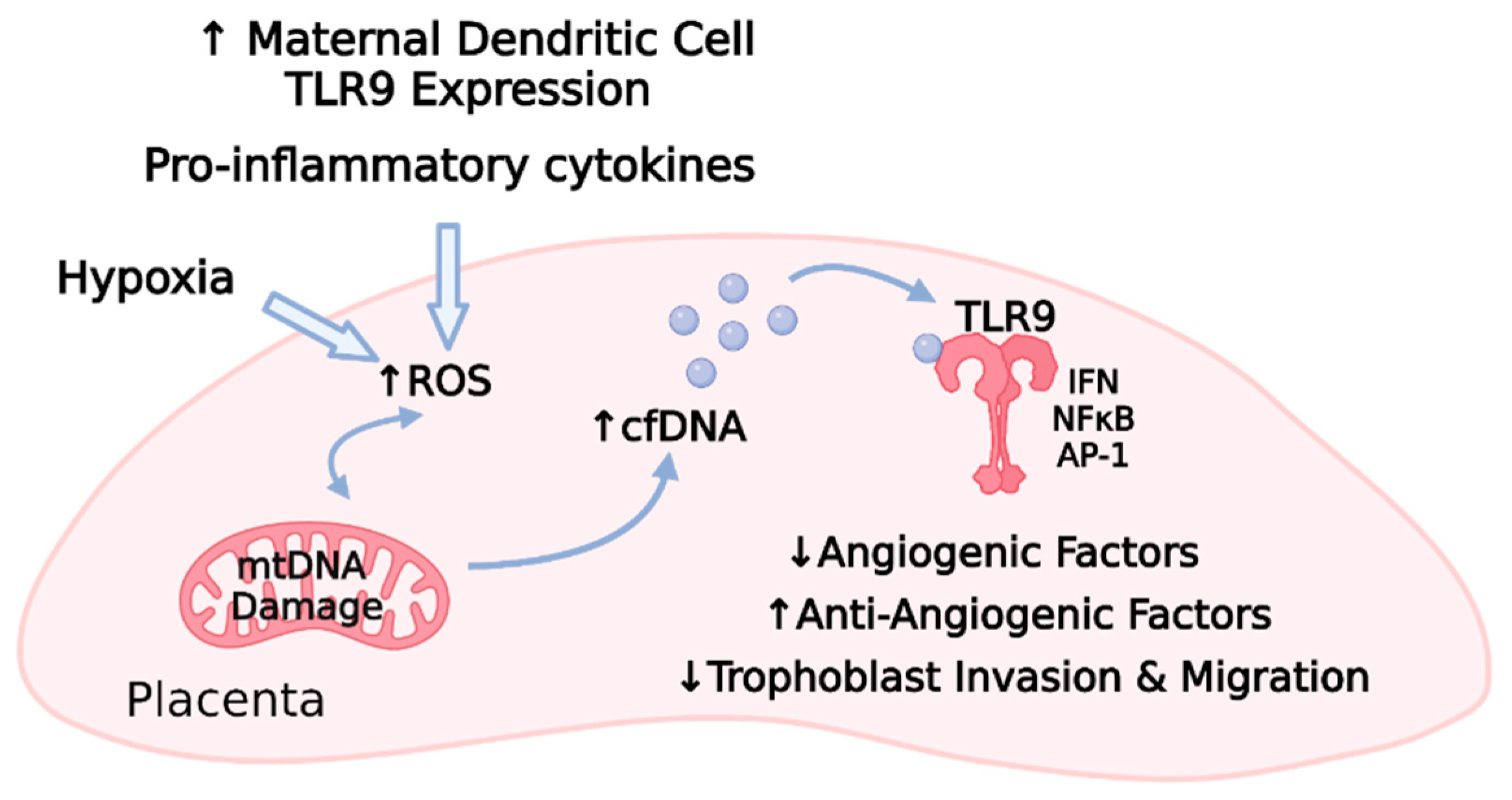
Figure 4. Maternal factors, including TLR9-induced proinflammatory cytokine release and placental hypoxia, promote a cascade of oxidative stress, mitochondrial DNA damage, cell-free DNA release, TLR9 activation, and subsequent TLR9 activation within the placenta [135][125][126][127][128][129][130][131][132][133][136][137]. Animal and cell culture models indicate that TLR9-related signaling results in decreased angiogenic factors, increased antiangiogenic factors, and impaired trophoblast function [135] (TLR9, toll-like receptor 9; ROS, reactive oxygen species; mtDNA, mitochondrial DNA; cfDNA, cell-free DNA; IFN, interferon; NFκB, nuclear factor kappa B; AP-1, activator protein-1; ↑ refers to upregulation; ↓ refers to downregulation).
References
- Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obs. Gynecol 2013, 122, 1122–1131.
- Rana, S.; Lemoine, E.; Granger, J.; Karumanchi, S.A. Preeclampsia: Pathophysiology, challenges, and perspectives. Circ. Res. 2019, 124, 1094–1112.
- Fox, R.; Kitt, J.; Leeson, P.; Aye, C.Y.L.; Lewandowski, A.J. Preeclampsia: Risk Factors, Diagnosis, Management, and the Cardiovascular Impact on the Offspring. J. Clin. Med. 2019, 8, 1625.
- Pankiewicz, K.; Szczerba, E.; Maciejewski, T.; Fijałkowska, A. Non-obstetric complications in preeclampsia. Prz. Menopauzalny 2019, 18, 99–109.
- Duley, L. The global impact of pre-eclampsia and eclampsia. Semin. Perinatol. 2009, 33, 130–137.
- Jeyabalan, A. Epidemiology of preeclampsia: Impact of obesity. Nutr. Rev. 2013, 71.
- Bellamy, L.; Casas, J.-P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ Clin. Res. Ed. 2007, 335, 974.
- Aykas, F.; Solak, Y.; Erden, A.; Bulut, K.; Dogan, S.; Sarli, B.; Acmaz, G.; Afsar, B.; Siriopol, D.; Covic, A.; et al. Persistence of cardiovascular risk factors in women with previous preeclampsia: A long-term follow-up study. J. Investig. Med. 2015, 63, 641–645.
- Ying, W.; Catov, J.M.; Ouyang, P. Hypertensive Disorders of Pregnancy and Future Maternal Cardiovascular Risk. J. Am. Heart Assoc. 2018, 7, e009382.
- Perez Botero, J.; Reese, J.A.; George, J.N.; McIntosh, J.J. Severe thrombocytopenia and microangiopathic hemolytic anemia in pregnancy: A guide for the consulting hematologist. Am. J. Hematol. 2021.
- Fingar, K.R.; Mabry-Hernandez, I.; Ngo-Metzger, Q.; Wolff, T.; Steiner, C.A.; Elixhauser, A. Delivery Hospitalizations Involving Preeclampsia and Eclampsia, 2005–2014: Statistical Brief #222. In Healthcare Cost and Utilization Project (HCUP) Statistical Briefs; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2006.
- Bell, M.J. A historical overview of preeclampsia-eclampsia. J. Obstet. Gynecol. Neonatal Nurs. JOGNN 2010, 39, 510–518.
- Högberg, U. The World Health Report 2005: “make every mother and child count”—including Africans. Scand. J. Public Health 2005, 33, 409–411.
- Ananth, C.V.; Keyes, K.M.; Wapner, R.J. Pre-eclampsia rates in the United States, 1980–2010: Age-period-cohort analysis. BMJ 2013, 347, f6564.
- Shih, T.; Peneva, D.; Xu, X.; Sutton, A.; Triche, E.; Ehrenkranz, R.A.; Paidas, M.; Stevens, W. The Rising Burden of Preeclampsia in the United States Impacts Both Maternal and Child Health. Am. J. Perinatol. 2016, 33, 329–338.
- Wallis, A.B.; Saftlas, A.F.; Hsia, J.; Atrash, H.K. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987–2004. Am. J. Hypertens 2008, 21, 521–526.
- Stevens, W.; Shih, T.; Incerti, D.; Ton, T.G.N.; Lee, H.C.; Peneva, D.; Macones, G.A.; Sibai, B.M.; Jena, A.B. Short-term costs of preeclampsia to the United States health care system. Am. J. Obs. Gynecol. 2017, 217, 237–248.
- Duckitt, K.; Harrington, D. Risk factors for pre-eclampsia at antenatal booking: Systematic review of controlled studies. BMJ 2005, 330, 565.
- Ghosh, G.; Grewal, J.; Männistö, T.; Mendola, P.; Chen, Z.; Xie, Y.; Laughon, S.K. Racial/ethnic differences in pregnancy-related hypertensive disease in nulliparous women. Ethn. Dis. 2014, 24, 283–289.
- Zhang, M.; Wan, P.; Ng, K.; Singh, K.; Cheng, T.H.; Velickovic, I.; Dalloul, M.; Wlody, D. Preeclampsia Among African American Pregnant Women: An Update on Prevalence, Complications, Etiology, and Biomarkers. Obs. Gynecol. Surv. 2020, 75, 111–120.
- Shahul, S.; Tung, A.; Minhaj, M.; Nizamuddin, J.; Wenger, J.; Mahmood, E.; Mueller, A.; Shaefi, S.; Scavone, B.; Kociol, R.D.; et al. Racial Disparities in Comorbidities, Complications, and Maternal and Fetal Outcomes in Women With Preeclampsia/eclampsia. Hypertens. Pregnancy 2015, 34, 506–515.
- Bezerra, P.C.; Leão, M.D.; Queiroz, J.W.; Melo, E.M.; Pereira, F.V.; Nóbrega, M.H.; Jeronimo, A.K.; Ferreira, L.C.; Jerônimo, S.M.; de Araújo, A.C. Family history of hypertension as an important risk factor for the development of severe preeclampsia. Acta Obs. Gynecol. Scand. 2010, 89, 612–617.
- Qiu, C.; Williams, M.A.; Leisenring, W.M.; Sorensen, T.K.; Frederick, I.O.; Dempsey, J.C.; Luthy, D.A. Family history of hypertension and type 2 diabetes in relation to preeclampsia risk. Hypertension 2003, 41, 408–413.
- Cho, G.J.; Jung, U.S.; Sim, J.Y.; Lee, Y.J.; Bae, N.Y.; Choi, H.J.; Park, J.H.; Kim, H.-J.; Oh, M.-J. Is preeclampsia itself a risk factor for the development of metabolic syndrome after delivery? Obs. Gynecol. Sci. 2019, 62, 233–241.
- Ryckman, K.K.; Borowski, K.S.; Parikh, N.I.; Saftlas, A.F. Pregnancy Complications and the Risk of Metabolic Syndrome for the Offspring. Curr. Cardiovasc. Risk Rep. 2013, 7, 217–223.
- Muijsers, H.E.C.; Roeleveld, N.; van der Heijden, O.W.H.; Maas, A.H.E.M. Consider Preeclampsia as a First Cardiovascular Event. Curr. Cardiovasc. Risk Rep. 2019, 13, 21.
- Lu, H.Q.; Hu, R. Lasting Effects of Intrauterine Exposure to Preeclampsia on Offspring and the Underlying Mechanism. AJP Rep. 2019, 9, e275–e291.
- Anderson, C.M. Preeclampsia: Exposing future cardiovascular risk in mothers and their children. J. Obs. Gynecol. Neonatal. Nurs. 2007, 36, 3–8.
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: The convergence point for multiple pathways. Am. J. Obstet. Gynecol. 2021.
- Roberts, J.M.; Rich-Edwards, J.W.; McElrath, T.F.; Garmire, L.; Myatt, L. Subtypes of Preeclampsia: Recognition and Determining Clinical Usefulness. Hypertension 2021, 77, 1430–1441.
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147.
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in Pathogenesis, Definitions, and Guidelines. Clin. J. Am. Soc. Nephrol. 2016, 11, 1102–1113.
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134–135, 1–10.
- Roberts, J.M.; Escudero, C. The placenta in preeclampsia. Pregnancy Hypertens. 2012, 2, 72–83.
- Fisher, S.J. Why is placentation abnormal in preeclampsia? Am. J. Obstet. Gynecol. 2015, 213, S115–S122.
- Garrido-Gomez, T.; Quiñonero, A.; Dominguez, F.; Rubert, L.; Perales, A.; Hajjar, K.A.; Simon, C. Preeclampsia: A defect in decidualization is associated with deficiency of Annexin A2. Am. J. Obs. Gynecol. 2020, 222, 376–376.e371.
- Garrido-Gomez, T.; Dominguez, F.; Quiñonero, A.; Diaz-Gimeno, P.; Kapidzic, M.; Gormley, M.; Ona, K.; Padilla-Iserte, P.; McMaster, M.; Genbacev, O.; et al. Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc. Natl. Acad. Sci. USA 2017, 114, E8468–E8477.
- Ng, S.-W.; Norwitz, G.A.; Pavlicev, M.; Tilburgs, T.; Simón, C.; Norwitz, E.R. Endometrial Decidualization: The Primary Driver of Pregnancy Health. Int. J. Mol. Sci. 2020, 21, 4092.
- Garrido-Gómez, T.; Castillo-Marco, N.; Cordero, T.; Simón, C. Decidualization resistance in the origin of preeclampsia. Am. J. Obs. Gynecol. 2020.
- Sánchez-Aranguren, L.C.; Prada, C.E.; Riaño-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372.
- Wang, A.; Rana, S.; Karumanchi, S.A. Preeclampsia: The role of angiogenic factors in its pathogenesis. Physiology 2009, 24, 147–158.
- Lee, E.S.; Oh, M.-J.; Jung, J.W.; Lim, J.-E.; Seol, H.-J.; Lee, K.-J.; Kim, H.-J. The levels of circulating vascular endothelial growth factor and soluble Flt-1 in pregnancies complicated by preeclampsia. J. Korean Med. Sci. 2007, 22, 94–98.
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658.
- Maynard, S.E.; Karumanchi, S.A. Angiogenic factors and preeclampsia. Semin. Nephrol. 2011, 31, 33–46.
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105.
- Smani, T.; Gómez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. Trp channels in angiogenesis and other endothelial functions. Front. Physiol. 2018, 9, 1731.
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264.
- Geva, E.; Ginzinger, D.G.; Zaloudek, C.J.; Moore, D.H.; Byrne, A.; Jaffe, R.B. Human placental vascular development: Vasculogenic and angiogenic (branching and nonbranching) transformation is regulated by vascular endothelial growth factor-a, angiopoietin-1, and angiopoietin-2. J. Clin. Endocrinol. Metab. 2002, 87, 4213–4224.
- Grummer, M.A.; Sullivan, J.A.; Magness, R.R.; Bird, I.M. Vascular endothelial growth factor acts through novel, pregnancy-enhanced receptor signalling pathways to stimulate endothelial nitric oxide synthase activity in uterine artery endothelial cells. Biochem. J. 2009, 417, 501–511.
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor–Associated Hypertension and Vascular Disease. Hypertension 2018, 71, E1–E8.
- Chau, K.; Hennessy, A.; Makris, A. Placental growth factor and pre-eclampsia. J. Hum. Hypertens. 2017, 31, 782–786.
- Williams, D.; Kenyon, A.; Adamson, D. Physiology. In Basic Science in Obstetrics and Gynaecology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 173–230.
- Dewerchin, M.; Carmeliet, P. PlGF: A multitasking cytokine with disease-restricted activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056.
- Lobmaier, S.M.; Figueras, F.; Mercade, I.; Crovetto, F.; Peguero, A.; Parra-Saavedra, M.; Ortiz, J.U.; Crispi, F.; Gratacós, E. Levels of maternal serum angiogenic factors in third-trimester normal pregnancies: Reference ranges, influence of maternal and pregnancy factors and fetoplacental Doppler indices. Fetal. Diagn. 2014, 36, 38–43.
- Ng, Q.J.; Han, J.Y.; Saffari, S.E.; Yeo, G.S.-H.; Chern, B.S.M.; Tan, K.H. Longitudinal circulating placental growth factor (PlGF) and soluble FMS-like tyrosine kinase-1 (sFlt-1) concentrations during pregnancy in Asian women: A prospective cohort study. BMJ Open 2019, 9, e028321.
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating Angiogenic Factors and the Risk of Preeclampsia. N. Engl. J. Med. 2004, 350, 672–683.
- Benschop, L.; Schalekamp-Timmermans, S.; Broere-Brown, Z.A.; Roeters van Lennep, J.E.; Jaddoe, V.W.V.; Roos-Hesselink, J.W.; Ikram, M.K.; Steegers, E.A.P.; Roberts, J.M.; Gandley, R.E. Placental Growth Factor as an Indicator of Maternal Cardiovascular Risk After Pregnancy. Circulation 2019, 139, 1698–1709.
- Roberts, J.M.; Rajakumar, A. Preeclampsia and soluble fms-like tyrosine kinase 1. J. Clin. Endocrinol. Metab. 2009, 94, 2252–2254.
- Thomas, C.P.; Andrews, J.I.; Raikwar, N.S.; Kelley, E.A.; Herse, F.; Dechend, R.; Golos, T.G.; Liu, K.Z. A recently evolved novel trophoblast-enriched secreted form of fms-like tyrosine kinase-1 variant is up-regulated in hypoxia and preeclampsia. J. Clin. Endocrinol. Metab. 2009, 94, 2524–2530.
- Adamson, S.L. sFLT1 in preeclampsia: Trophoblast defense against a decidual VEGFA barrage? J. Clin. Investig. 2014, 124, 4690–4692.
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22.
- Sela, S.; Itin, A.; Natanson-Yaron, S.; Greenfield, C.; Goldman-Wohl, D.; Yagel, S.; Keshet, E. A novel human-specific soluble vascular endothelial growth factor receptor 1: Cell type-specific splicing and implications to vascular endothelial growth factor homeostasis and preeclampsia. Circ. Res. 2008, 102, 1566–1574.
- Nagamatsu, T.; Fujii, T.; Kusumi, M.; Zou, L.; Yamashita, T.; Osuga, Y.; Momoeda, M.; Kozuma, S.; Taketani, Y. Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: An implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology 2004, 145, 4838–4845.
- Li, H.; Gu, B.; Zhang, Y.; Lewis, D.F.; Wang, Y. Hypoxia-induced increase in soluble Flt-1 production correlates with enhanced oxidative stress in trophoblast cells from the human placenta. Placenta 2005, 26, 210–217.
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606.
- Oujo, B.; Perez-Barriocanal, F.; Bernabeu, C.; Lopez-Novoa, J. Membrane and Soluble Forms of Endoglin in Preeclampsia. Curr. Mol. Med. 2013, 13, 1345–1357.
- Nassiri, F.; Cusimano, M.D.; Scheithauer, B.W.; Rotondo, F.; Fazio, A.; Yousef, G.M.; Syro, L.V.; Kovacs, K.; Lloyd, R.V. Endoglin (CD105): A review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res. 2011, 31, 2283–2290.
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.I.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649.
- St-Jacques, S.; Forte, M.; Lye, S.J.; Letarte, M. Localization of endoglin, a transforming growth factor-β binding protein, and of CD44 and integrins in placenta during the first trimester of pregnancy. Biol. Reprod. 1994, 51, 405–413.
- Alam, S.; Ahmad, S.; Zafeer, F.; Rizvi, A.A.; Gulati, R.; Rashid, S. Role of TGF-Β1 in The Pathogenesis of Pre-Eclampsia. Ann. Int. Med. Dent. Res. 2017, 3, 1.
- Caniggia, I.; Taylor, C.V.; Ritchie, J.W.K.; Lye, S.J.; Letarte, M. Endoglin regulates trophoblast differentiation along the invasive pathway in human placental villous explants. Endocrinology 1997, 138, 4977–4988.
- Ayatollahi, M.; Geramizadeh, B.; Samsami, A. Transforming growth factor beta-1 influence on fetal allografts during pregnancy. Transplant. Proc. 2005, 37, 4603–4604.
- Nikuei, P.; Rajaei, M.; Malekzadeh, K.; Nejatizadeh, A.; Mohseni, F.; Atashabparvar, A. Accuracy of soluble endoglin for diagnosis of preeclampsia and its severity. Iran. Biomed. J. 2017, 21, 312–320.
- Powe, C.E.; Levine, R.J.; Karumanchi, S.A. Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011, 123, 2856–2869.
- Bell, M.J.; Roberts, J.M.; Founds, S.A.; Jeyabalan, A.; Terhorst, L.; Conley, Y.P. Variation in endoglin pathway genes is associated with preeclampsia: A case-control candidate gene association study. BMC Pregnancy Childbirth 2013, 13.
- Jerkic, M.; Rivas-Elena, J.V.; Prieto, M.; Carrón, R.; Sanz-Rodríguez, F.; Pérez-Barriocanal, F.; Rodríguez-Barbero, A.; Bernabéu, C.; López-Novoa, J.M. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004, 18, 609–611.
- Mano, Y.; Kotani, T.; Shibata, K.; Matsumura, H.; Tsuda, H.; Sumigama, S.; Yamamoto, E.; Iwase, A.; Senga, T.; Kikkawa, F. The loss of endoglin promotes the invasion of extravillous trophoblasts. Endocrinology 2011, 152, 4386–4394.
- Williams, P.J.; Mistry, H.D.; Innes, B.A.; Bulmer, J.N.; Broughton Pipkin, F. Expression of AT1R, AT2R and AT4R and Their Roles in Extravillous Trophoblast Invasion in the Human. Placenta 2010, 31, 448–455.
- Guo, D.F.; Sun, Y.L.; Hamet, P.; Inagami, T. The angiotensin II type 1 receptor and receptor-associated proteins. Cell Res. 2001, 11, 165–180.
- LaMarca, B. Endothelial dysfunction. An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva Ginecol. 2012, 64, 309–320.
- LaMarca, B.; Wallukat, G.; Llinas, M.; Herse, F.; Dechend, R.; Granger, J.P. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension 2008, 52, 1168–1172.
- LaMarca, B.; Parrish, M.; Ray, L.F.; Murphy, S.R.; Roberts, L.; Glover, P.; Wallukat, G.; Wenzel, K.; Cockrell, K.; Martin, J.N., Jr.; et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: Role of endothelin-1. Hypertension 2009, 54, 905–909.
- Parrish, M.R.; Murphy, S.R.; Rutland, S.; Wallace, K.; Wenzel, K.; Wallukat, G.; Keiser, S.; Ray, L.F.; Dechend, R.; Martin, J.N.; et al. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am. J. Hypertens. 2010, 23, 911–916.
- Bezerra Maia E Holanda Moura, S.; Marques Lopes, L.; Murthi, P.; da Silva Costa, F. Prevention of preeclampsia. J. Pregnancy 2012, 2012, 435090.
- Brown, C.M.; Garovic, V.D. Drug treatment of hypertension in pregnancy. Drugs 2014, 74, 283–296.
- Duhig, K.; Vandermolen, B.; Shennan, A. Recent advances in the diagnosis and management of pre-eclampsia. F1000Res 2018, 7, 242.
- Spradley, F.T.; Tan, A.Y.; Joo, W.S.; Daniels, G.; Kussie, P.; Karumanchi, S.A.; Granger, J.P. Placental growth factor administration abolishes placental ischemia-induced hypertension. Hypertension 2016, 67, 740–747.
- Zhu, M.; Ren, Z.; Possomato-Vieira, J.S.; Khalil, R.A. Restoring placental growth factor-soluble fms-like tyrosine kinase-1 balance reverses vascular hyper-reactivity and hypertension in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R505–R521.
- Thadhani, R.; Hagmann, H.; Schaarschmidt, W.; Roth, B.; Cingoez, T.; Karumanchi, S.A.; Wenger, J.; Lucchesi, K.J.; Tamez, H.; Lindner, T.; et al. Removal of soluble fms-like tyrosine kinase-1 by dextran sulfate apheresis in preeclampsia. J. Am. Soc. Nephrol. 2016, 27, 903–913.
- Bridges, J.P.; Gilbert, J.S.; Colson, D.; Gilbert, S.A.; Dukes, M.P.; Ryan, M.J.; Granger, J.P. Oxidative stress contributes to soluble fms-like tyrosine kinase-1 induced vascular dysfunction in pregnant rats. Am. J. Hypertens. 2009, 22, 564–568.
- Félétou, M. The Endothelium: Part 1: Multiple Functions of the Endothelial Cells- Focus on Endothelium-Derived Vasoactive Mediators. In Colloquium Series on Integrated Systems Physiology; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011.
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113.
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469.
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms regulating endothelial permeability. Pulm. Circ. 2014, 4, 535–551.
- Tseng, E.; Yee Teoh, S.S.; Wang, Y.; Nie, G. Elevated protease HtrA4 in the maternal circulation of preeclampsia may contribute to endothelial barrier disruption by cleaving key junctional protein VE-cadherin. Placenta 2019, 76, 51–53.
- Wang, Y.; Lewis, D.F.; Alexander, J.S.; Granger, D.N. Endothelial barrier function in preeclampsia. Front. Biosci. 2007, 12, 2412–2424.
- Hayman, R.; Warren, A.; Brockelsby, J.; Johnson, I.; Baker, P. Plasma from women with pre-eclampsia induces an in vitro alteration in the endothelium-dependent behaviour of myometrial resistance arteries. BJOG Int. J. Obstet. Gynaecol. 2000, 107, 108–115.
- McIntosh, J.J.; Derayunan, A.; Hader, S.; Lohr, N.; Beyer, A.; Gutterman, D. Impaired Microvascular Endothelial Function in Preeclampsia. FASEB J. 2020, 34, 1.
- Chambers, J.C.; Fusi, L.; Malik, I.S.; Haskard, D.O.; De Swiet, M.; Kooner, J.S. Association of maternal endothelial dysfunction with preeclampsia. J. Am. Med. Assoc. 2001, 285, 1607–1612.
- Sandgren, J.A.; Deng, G.; Linggonegoro, D.W.; Scroggins, S.M.; Perschbacher, K.J.; Nair, A.R.; Nishimura, T.E.; Zhang, S.Y.; Agbor, L.N.; Wu, J.; et al. Arginine vasopressin infusion is sufficient to model clinical features of preeclampsia in mice. JCI Insight 2018, 3, e99403.
- Vanwijk, M.J.; Kublickiene, K.; Boer, K.; VanBavel, E. Vascular function in preeclampsia. Cardiovasc. Res. 2000, 47, 38–48.
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496.
- Can, M.; Guven, B.; Bektas, S.; Arikan, I. Oxidative stress and apoptosis in preeclampsia. Tissue Cell 2014, 46, 477–481.
- Cornelius, D.C. Preeclampsia: From inflammation to immunoregulation. Clin. Med. Insights Blood Disord. 2018, 11.
- Toniolo, A.; Buccellati, C.; Pinna, C.; Gaion, R.M.; Sala, A.; Bolego, C. Cyclooxygenase-1 and prostacyclin production by endothelial cells in the presence of mild oxidative stress. PLoS ONE 2013, 8, e56683.
- Brock, T.G.; McNish, R.W.; Peters-Golden, M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J. Biol. Chem. 1999, 274, 11660–11666.
- Walsh, S.W. Preeclampsia: An imbalance in placental prostacyclin and thromboxane Production. Am. J. Obstet. Gynecol. 1985, 152, 335–340.
- Atallah, A.; Lecarpentier, E.; Goffinet, F.; Doret-Dion, M.; Gaucherand, P.; Tsatsaris, V. Aspirin for Prevention of Preeclampsia. Drugs 2017, 77, 1819–1831.
- Lewis, D.F.; Canzoneri, B.J.; Gu, Y.; Zhao, S.; Wang, Y. Maternal Levels of Prostacyclin, Thromboxane, ICAM, and VCAM in Normal and Preeclamptic Pregnancies. Am. J. Reprod. Immunol. 2010, 64, 376–383.
- Walsh, S.W. Low-Dose Aspirin: Treatment for the Imbalance of Increased Thromboxane and Decreased Prostacyclin in Preeclampsia. Am. J. Perinatol. 1989, 6, 124–132.
- Tannetta, D.S.; Hunt, K.; Jones, C.I.; Davidson, N.; Coxon, C.H.; Ferguson, D.; Redman, C.W.; Gibbins, J.M.; Sargent, I.L.; Tucker, K.L. Syncytiotrophoblast Extracellular Vesicles from Pre-Eclampsia Placentas Differentially Affect Platelet Function. PLoS ONE 2015, 10, e0142538.
- Duley, L.; Meher, S.; Hunter, K.E.; Seidler, A.L.; Askie, L.M. Antiplatelet agents for preventing pre-eclampsia and its complications. Cochrane Database Syst. Rev. 2007, CD004659.
- ACOG Committee Opinion No. 743: Low-Dose Aspirin Use During Pregnancy. Obs. Gynecol 2018, 132, e44–e52.
- Periayah, M.H.; Halim, A.S.; Saad, A.Z.M. Mechanism action of platelets and crucial blood coagulation pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327.
- Jakobsen, C.; Larsen, J.B.; Fuglsang, J.; Hvas, A.M. Platelet function in preeclampsia–a systematic review and meta-analysis. Platelets 2019, 30, 549–562.
- Goksu Erol, A.Y.; Nazli, M.; Yildiz, S.E. Significance of platelet endothelial cell adhesion molecule-1 (PECAM-1) and intercellular adhesion molecule-1 (ICAM-1) expressions in preeclamptic placentae. Endocrine 2012, 42, 125–131.
- Reese, J.A.; Peck, J.D.; Deschamps, D.R.; McIntosh, J.J.; Knudtson, E.J.; Terrell, D.R.; Vesely, S.K.; George, J.N. Platelet counts during pregnancy. N. Engl. J. Med. 2018, 379, 32–43.
- Reese, J.A.; Peck, J.D.; Yu, Z.; Scordino, T.A.; Deschamps, D.R.; McIntosh, J.J.; Terrell, D.R.; Vesely, S.K.; George, J.N. Platelet sequestration and consumption in the placental intervillous space contribute to lower platelet counts during pregnancy. Am. J. Hematol. 2019, 94, E8–E11.
- Cines, D.B.; Levine, L.D. Thrombocytopenia in pregnancy. Blood 2017, 130, 2271–2277.
- Ciobanu, A.M.; Colibaba, S.; Cimpoca, B.; Peltecu, G.; Panaitescu, A.M. Thrombocytopenia in Pregnancy. Maedica 2016, 11, 55–60.
- Heilmann, L.; Rath, W.; Pollow, K. Hemostatic abnormalities in patients with severe preeclampsia. Clin. Appl. Thromb. Hemost. 2007, 13, 285–291.
- Han, L.; Liu, X.; Li, H.; Zou, J.; Yang, Z.; Han, J.; Huang, W.; Yu, L.; Zheng, Y.; Li, L. Blood coagulation parameters and platelet indices: Changes in normal and preeclamptic pregnancies and predictive values for preeclampsia. PLoS ONE 2014, 9, e114488.
- Youssef, L.; Miranda, J.; Blasco, M.; Paules, C.; Crovetto, F.; Palomo, M.; Torramade-Moix, S.; García-Calderó, H.; Tura-Ceide, O.; Dantas, A.P.; et al. Complement and coagulation cascades activation is the main pathophysiological pathway in early-onset severe preeclampsia revealed by maternal proteomics. Sci. Rep. 2021, 11, 3048.
- Higgins, J.R.; Bonnar, J.; Norris, L.A.; Darling, M.R.; Walshe, J.J. The effect of pre-eclampsia on coagulation and fibrinolytic activation in the neonate. Thromb. Res. 2000, 99, 567–570.
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548.
- Tenório, M.B.; Ferreira, R.C.; Moura, F.A.; Bueno, N.B.; de Oliveira, A.C.M.; Goulart, M.O.F. Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia. Oxidative Med. Cell. Longev. 2019, 2019, 8238727.
- Walsh, S.W. Plasma from preeclamptic women stimulates transendothelial migration of neutrophils. Reprod. Sci. 2009, 16, 320–325.
- Williamson, R.D.; McCarthy, F.P.; Kenny, L.C.; McCarthy, C.M. Activation of a TLR9 mediated innate immune response in preeclampsia. Sci. Rep. 2019, 9, 1–8.
- Lemasters, J.J.; Nieminen, A.L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim. Et Biophys. Acta Bioenerg. 1998, 1366, 177–196.
- Van Houten, B.; Hunter, S.E.; Meyer, J.N. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front. Biosci. 2016, 21, 42–54.
- Thurairajah, K.; Briggs, G.D.; Balogh, Z.J. The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies. Eur. J. Trauma Emerg. Surg. 2018, 44, 325–334.
- Chockalingam, A.; Brooks, J.C.; Cameron, J.L.; Blum, L.K.; Leifer, C.A. TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol. Cell Biol. 2009, 87, 209–217.
- Goulopoulou, S.; Matsumoto, T.; Bomfim, G.F.; Webb, R.C. Toll-like receptor 9 activation: A novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin. Sci. 2012, 123, 429–435.
- Bao, W.; Xia, H.; Liang, Y.; Ye, Y.; Lu, Y.; Xu, X.; Duan, A.; He, J.; Chen, Z.; Wu, Y.; et al. Toll-like Receptor 9 Can be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci. Rep. 2016, 6, 22579.
- He, B.; Yang, X.; Li, Y.; Huang, D.; Xu, X.; Yang, W.; Dai, Y.; Zhang, H.; Chen, Z.; Cheng, W. TLR9 (toll-like receptor 9) agonist suppresses angiogenesis by differentially regulating VEGFA (Vascular Endothelial Growth Factor A) and sFLT1 (Soluble Vascular Endothelial Growth Factor Receptor 1) in Preeclampsia. Hypertension 2018, 71, 671–680.
- Marschalek, J.; Wohlrab, P.; Ott, J.; Wojta, J.; Speidl, W.; Klein, K.U.; Kiss, H.; Pateisky, P.; Zeisler, H.; Kuessel, L. Maternal serum mitochondrial DNA (mtDNA) levels are elevated in preeclampsia–a matched case-control study. Pregnancy Hypertens. 2018, 14, 195–199.
- Panda, B.; Panda, A.; Ueda, I.; Abrahams, V.M.; Norwitz, E.R.; Stanic, A.K.; Young, B.C.; Ecker, J.L.; Altfeld, M.; Shaw, A.C.; et al. Dendritic cells in the circulation of women with preeclampsia demonstrate a pro-inflammatory bias secondary to dysregulation of TLR receptors. J. Reprod. Immunol. 2012, 94, 210–215.
More
Information
Subjects:
Obstetrics & Gynaecology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
17 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No