Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Katarzyna Sobierajska | + 2296 word(s) | 2296 | 2021-11-02 06:54:43 | | | |
| 2 | Camila Xu | Meta information modification | 2296 | 2021-11-16 08:39:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sobierajska, K. Cytoskeleton Reorganization in EndMT. Encyclopedia. Available online: https://encyclopedia.pub/entry/16033 (accessed on 26 July 2026).
Sobierajska K. Cytoskeleton Reorganization in EndMT. Encyclopedia. Available at: https://encyclopedia.pub/entry/16033. Accessed July 26, 2026.
Sobierajska, Katarzyna. "Cytoskeleton Reorganization in EndMT" Encyclopedia, https://encyclopedia.pub/entry/16033 (accessed July 26, 2026).
Sobierajska, K. (2021, November 16). Cytoskeleton Reorganization in EndMT. In Encyclopedia. https://encyclopedia.pub/entry/16033
Sobierajska, Katarzyna. "Cytoskeleton Reorganization in EndMT." Encyclopedia. Web. 16 November, 2021.
Copy Citation
EndMT-derived cells, known as the myofibroblasts or cancer-associated fibroblasts (CAFs), are characterized by the loss of cell–cell junctions, loss of endothelial markers, and gain in mesenchymal ones.
EndMT
cancer
fibrosis
CAFs
cytoskeleton
myofibroblasts
1. Introduction
The vascular endothelium is composed of a monolayer of tightly connected endothelial cells that cover the interior surface of blood vessels. It is not only the barrier located between circulating blood and tissues but also a vital organ involved in numerous functions. Under physiological conditions, the endothelium maintains cardiovascular homeostasis via a wide range of biologically active substances such as cytokines, chemokines, and growth factors [1]. It is mainly involved in the regulation of vascular tone, fluid homeostasis, and host defense [2]. Unfortunately, chronic inflammation, oxidative stress, and shear stress cause disorders of endothelium function that induce procoagulant properties, leading to severe sepsis. They are also responsible for sickle cell disease, macular degeneration, prematurity, or diabetic retinopathy [3]. Endothelial dysfunction leads to atherosclerotic lesions and, consequently, an increased risk of cardiovascular events such as idiopathic pulmonary arterial hypertension, stroke, infarction, or heart failure [4]. There is a growing amount of evidence that endothelial cells can serve as sources of myofibroblasts in fibrosis, such as cystic, kidney, heart, dermal, pulmonary, and intestinal fibrosis, as well as cancer-associated fibroblasts (CAFs) in neoplasia [5][6][7][8][9][10][11][12][13][14][15].
It has been estimated that around 45% of natural deaths yearly can be associated with different fibrotic disorders in the USA [16]. Organ fibrosis is a common pathological state of slightly known etiology, resulting in chronic tissue injury defined as an increasing production and deposition of extracellular matrix (ECM) components [17]. Chronic inflammation causes fibrosis tissue to recruit numerous activated fibroblasts called myofibroblasts. The myofibroblast was initially described in the granulation tissue of wound healing as the cells with prominent cytoplasmic microfilament bundles and peripheral focal adhesions. Electron microscopy revealed that myofibroblasts characterized the abundant expression of α-SMA, the isoform of actin specific for smooth muscle cells [18]. Myofibroblasts are a heterogeneous group of cells with a comprehensive source of cells, including fibroblasts, circulating bone marrow-derived cells, and epithelial or endothelial cells [19]. They are characterized as α-SMA-positive myofibroblasts, which are the principal source of the enormous extracellular matrix (ECM), including collagen type I, fibronectin, hyaluronan, and elastin [19][20][21]. In turn, cancer-associated fibroblasts (CAFs), a type of activated fibroblast located in the cancer niche, are cells with significant heterogeneity and plasticity. CAFs are widely described as the leading promoter of tumor progression and metastasis. It should be mentioned that CAFs may also have certain tumor-suppressive functions in the early stage of tumors [22].
Fibrosis and neoplastic disease are two distinct and distant disease entities. However, they have a common denominator: the excessive deposition of ECM proteins by myofibroblasts/CAFs, which changes the homeostasis within the niche. One of the well-known sources of myofibroblasts/CAFs is endothelial cells transdifferentiated into them by the endothelial–mesenchymal transition (Figure 1).
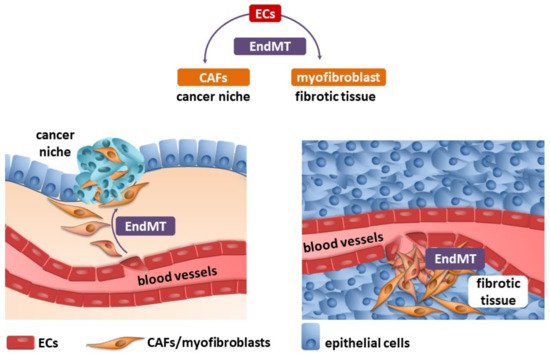
Figure 1. Endothelial cells (ECs) undergo an endothelial–mesenchymal transition (EndMT), which are the source of cancer-associated fibroblasts (CAFs) and myofibroblasts (detailed description in the text).
2. Endothelial–Mesenchymal Transition
The endothelial–mesenchymal transition (EndMT) was initially observed during heart development [23][24]. Several inflammatory mediators, including pro-inflammatory cytokines (e.g., interleukin 1-β, IL 1-β; tumor necrosis factor-α, TNF-α), growth factors (e.g., fetal grown factor, FGF), oxidative stress, shear stress, and toxins, induce the conversion of endothelial cells into mesenchymal fibroblast-like cells that promote disease progression [25][26] (Figure 2, Table 1). EndMT appears to be regulated by complex molecular mechanisms and different signaling pathways. Members of the Tumor Growth Factor-β (TGF-β) family are the most known inducers of EndMT. Three TGF-β members, TGF-β1, TGF-β2, and TGF-β3, act through TGF-β type I and II receptors to form multimeric complexes and subsequently launch the downstream Smad-dependent and Smad-independent signaling pathways [19]. The Smad-dependent signaling pathway is essential for increasing the expression of cell-adhesion-suppressing zinc-finger transcription factors (TF) such as Snail, Twist, Zeb, and Slug [27][28]. In the canonical pathway, induction of TGF-β receptors causes activation of the TGF-β type I receptor that binds and phosphorylates Smad2/3, which interacts with Smad4 to form a transcription complex that translocates to the nucleus and triggers the expression of the genes mentioned above [19]. In addition, certain TGF-β family members (TGF-β2, BMP2, and BMP4) induce EndMT by signaling through the TGF-β type II receptor (TGFBR2) [19]. The in vivo relevance of this mechanism is illustrated by the EC-derived heterotopic ossification observed in patients with fibrodysplasia ossificans progressiva, which is due to an overactive mutant TGFBR2 [29]. The crucial role of the TGF-β super-family in the EndMT induction has been validated in cell line studies and in vivo mice experiments. The knockdown and knockout of several TGF-β signaling-related genes, such as Smad2, Smad3, and TGFBR2, prevented EndMT [29].
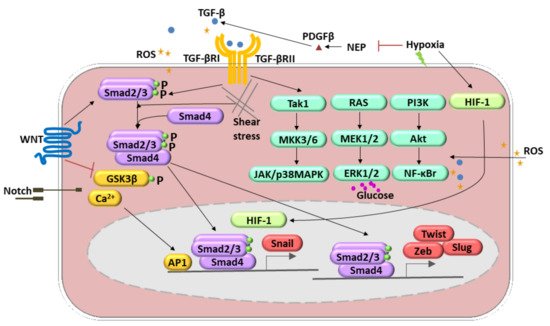
Figure 2. Molecular pathways involved in the endothelial-to-mesenchymal transition (EndMT) regulation. These include transforming growth factor (TGF)-β, TNF-α, BMP, FGF, IL-1 β, Notch, WNT signaling, oxidative stress, and shear stress. TGF-β-induced EndMT involves the canonical Smad2/3 pathway. Additionally, it can activate Smad-independent pathways (1. Tak1, MKK3/6, JAK/p38MAPK; 2. RAS, MEK1/2, ERK1/2; 3. PI3K, Akt, NF-κB). Hypoxia induces EndMT through the effects of HIF-1α activation of Snail1. Shear stress forces (represented by undulating arrows) induce EndMT through several different molecular mechanisms. Other mechanisms include reactive oxygen species (ROS) generation and activation of NF-κB followed by the PI3K, resulting in the increased production and accumulation of ROS (yellow stars). A high level of glucose acts as an ERK1/2 activator and, therefore, regulates the EndMT.
Several other signaling pathways are involved in the induction of the EndMT. The Wnt signal transduction is run via Smad-dependent TGF-β signaling, and canonical (i.e., involving β-catenin) and noncanonical Wnt signaling pathways [30], whereas Notch pathway activation resulted in Snail upregulation [31]. Additionally, it has been shown that Kaposi’s sarcoma-associated herpesvirus causes EndMT via Notch signaling independently of the TGF-β pathway [32]. Within noncanonical pathways, Rho-GTPase-actin and Smad-independent signaling pathways such as Akt/NF-ĸB and MAPK/ERK can be recognized [33][34][35][36] (Figure 2). Recent evidence also suggests that small RNAs, particularly microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), are crucial mediators of EndMT [37]. Hypoxia is the condition able to induce the EndMT. These molecular pathways engage Snail and hypoxia-inducible factor-1 α (HIF-1α) upregulation. Those processes were observed in radiation-induced pulmonary fibrosis [7]. HIF-1 may also decrease neprilysin (NEP) and, in this way, upregulated platelet-derived growth factor (PDGF)-β) and stimulation of TGF-β1 signaling [38]. Reactive oxygen species (ROS) works by differential pathways. They are able to induce TGF-β expression and, therefore, via a positive feedback loop, lead to ROS production. Secondly, ROS might act as activators of nuclear factor-κΒ (NF-κB) signaling that stimulate EndMT synergistically with TGF-β [39]. Moreover, one of the crucial enzymes responsible for ROS production, NADPH oxidase 4 (NOX4), mediated the TGF-β-dependent production of myofibroblasts by EndMT [40].
Table 1. Molecular pathways involved in EndMT.
| EndMT Inductor | Receptors | Molecular Pathways | Refereces |
|---|---|---|---|
| TGF-β1, TGF-β2, TGF-β3 |
TGFBR1, TGFBR2 |
Smad-dependent pathways (Smad2, Smad3, and Smad4) | [27][28] |
| TGF-β1, TGF-β2, TGF-β3 |
TGFBR1, TGFBR2 |
Smad-independent pathways include the mitogen-activated protein kinase (MAPK) family of serine/threonine-specific protein kinases, phosphatidylinositol 3-kinase (PI3K), RhoA, Rac, c-Abl, and protein kinase C (PKC)-δ. MAPK pathways: extracellular signal-regulated kinase (ERK), p38 MAPK, and c-Jun NH2-terminal kinases (JNK). | [33][34][35][36] |
| Notch | NOTCHR | Notch and TGF-β synergistically stimulate Snail expression | [31] |
| Notch | NOTCHIR | GSKβ inhibiton, calcium ions upregulation | [33] |
| Wnt | Frizzed | Smad-dependent pathways (Smad2, Smad3, and Smad4) | [30] |
| Wnt | Frizzed | GSKβ inhibiton | [30] |
| HIF-1 | Neprilysin downregulation induces upregulation of PDGF-β and finally TGF-β1 signaling induction (hypoxia) | [38] | |
| ROS | TGFBR1, TGFBR2 | TGF-β expression resulted in ROS production in the positive loop | [39] |
| ROS | Induction of NF-κB signaling that, with TGF-β pathway, stimulated EndmT | [39] | |
| NOX4 | ROS production caused TGF-β pathway induction | [40] | |
| Shear stress | High shear stress via ERK5 inhibits EndMT | [41] | |
| Shear stress | Cyclic strain, caused by a perpendicular stretching force on the vessel wall, has been shown to potentiate EndMT by augmenting both TGF-β and Wnt signaling | [42][43] | |
| High glucose | ERK1/2 phosphorylation | [44] |
Shear stress that ensures homeostasis of ECs can modulate the EndMT via TGF-β-dependent signaling independent of its intensity. The in vivo and organ-in-chip experiments have shown that high shear stress appears to inhibit EndMT [41] via extracellular-signal-regulated kinase 5 (ERK5), whereas ERK5 overactivation prevents the EndMT in cells exposed to a disturbed flow or are stimulated by TGF-β under static conditions [45]. Different mechanical stresses, termed cyclic strain, and caused by a perpendicular stretching force on the vessel wall, have been shown to potentiate the EndMT by augmenting both TGF-β and Wnt signaling [42][43]. High glucose concentrations have been shown to cause EndMT, involving extracellular signal-regulated kinase (ERK) 1/2 phosphorylation.
During EndMT, endothelial cells lose their typical phenotype and acquire mesenchymal features, characterized by a shift in endothelial markers toward mesenchymal ones (Figure 3). However, identifying CAFs, it should be remembered that despite many CAF biomarkers, none of them are specific to CAFs [44]. Their expression depends on the cancer type and probably the organ undergoing fibrosis. The endothelial markers commonly include PECAM (CD31), von Willebrand factor (vWF), VE-cadherin (CD144), tyrosine kinase with immunoglobulin-like and EGF-like domains 1 and 2 (TIE1 and TIE2), eNOS, and platelet-derived growth factor (PDGF). To confirm the occurrence of the EndMT, the most often detected mesenchymal markers include α-smooth muscle actin (α-SMA), N-cadherin, calponin, fibroblast-specific protein-1 (FSP-1), vimentin, fibronectin (FN), collagen types I and III, and matrix metalloproteinase 2 and 9 (MMP-2 and MMP-9, respectively). It should be mentioned that other frequently used typical EMT markers, such as E-cadherin, claudin, occludin, and cytokeratin, are not usually used in studies of EndMT [46][47].
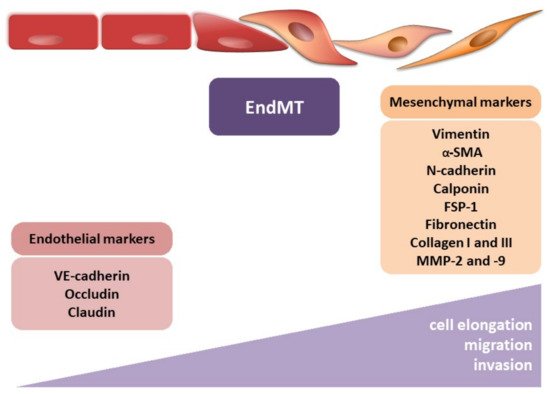
Figure 3. Endothelial–mesenchymal transition. The figure illustrates the morphological modulation (cell elongation) and increasing migration and invasion ability of mesenchymal cells. The observed phenotypic alterations are accompanied by changes in gene expression, i.e., decrease in endothelial markers (VE-cadherin, occludin, and claudin) and increase in mesenchymal markers (vimentin, α-smooth muscle actin (α-SMA), N-cadherin, calponin, fibroblast-specific protein-1 (FSP-1), fibronectin, collagen I, collagen III, metalloproteinase-2 (MMP-2), and metalloproteinase-9 (MMP-9)).
Next to molecular modulation, EndMT is manifested by profound morphological and functional changes. ECs that undergo EndMT are characterized by a phenotypic switch involving a loss of cellular adhesion (Figure 4) due to the downregulation of proteins involved in cell–cell junctions (adherens junction (AJs) and tight junctions (TJs)) and cytoskeletal reorganization, which converts tightly compacted cobblestone-like cells into spindle-shaped cells with no apical–basal polarity. EndMT-derived cells thus exhibit an enhanced migratory potential and increased extracellular matrix production, both of which are hallmarks of invasive cells [48] (Figure 4).
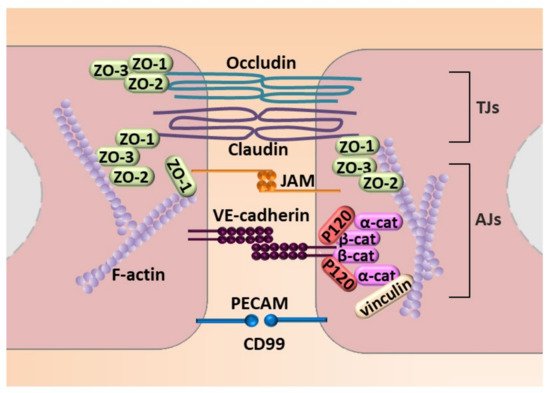
Figure 4. Molecular mechanisms engaged in endothelial cell–cell junction. The figure illustrates adhesion molecules that regulate AJs that consist of VE-cadherin with zonula occludes-1, -2, and -3 (ZO-1, ZO-2, and ZO-3) that interact with a complex of P-120, and a α-cathenin and β-cathenin (α-cat and β-cat, respectively) complex that connects VE-cadherin to stress fibers created by fibrillar actin (F-actin). TJs are created by occludin and claudin and JAM that interacts with ZO-1, ZO-2, and ZO-3. An additional element of cell–cell contact is the PECAM.
Such a reorganization of the cytoskeleton converts EC’s apicobasal polarity toward a front-end back polarity to form spindle-shaped cells with enhanced properties of migration [48] (Figure 3). As a consequence of the processes mentioned above and the influence of the effectors, cell–cell connections are disrupted, and the pro-migratory, pro-inflammatory, and pro-secretory abilities of the cells are increased. That chronic state contributes to the development of fibrotic and neoplastic diseases.
3. Cytoskeleton in Endothelial–Mesenchymal Transition
A cellular cytoskeleton is a network of fibrous protein structures forming a scaffold for cell organelles. It plays an essential role in maintaining cell shape, membrane dynamics, intracellular transport, organelle positioning, cell polarization, movement, and division. Three types of structures constitute the basis of the cytoskeleton: intermediate filaments, microtubules (MT), and actin cytoskeleton (microfilaments) [49]. Together with associated proteins regulating their polymerization, motor proteins, and proteins connecting individual elements of the cytoskeleton, these filaments form a functional structure.
The first type of biopolymer is intermediate filaments responsible for ensuring the stability of the cell structure. The primary role of the second type of filament, microtubules, is to counteract mechanical stresses, transport intracellular organelles, as well as build the karyokinetic spindle. The third type of biopolymer is the actin cytoskeleton (microfilaments), which is mainly responsible for cell movement but also cell adhesion and migration [49].
The interrelationship between the cytoskeleton elements and participation in the transmission of environmental stimuli make the cytoskeleton play a key role in the proper development and functioning of tissues and organs, and thus in maintaining homeostasis of the organism [50]. The cytoskeleton is a dynamic system in which both the exchange of subunits in the already existing polymers and the local or global reconstruction of the fiber network take place (possible through the processes of polymerization and depolymerization of individual filaments) [51]. Such cytoskeleton dynamics enables the elimination of damaged subunits or fragments of structures and their replacement by proper units and the reorganization of the cytoskeleton in response to various internal and external stimuli, e.g., during cell division or differentiation and cell migration. Disorders in the proper functioning of the cytoskeleton lead to many serious diseases (e.g., myopathy, neuropathy, ciliopathy, and neoplastic diseases) [52].
4. EndMT Inhibition in Fibrosis and Cancer Treatment
As has already been discussed, EndMT is instrumental in the pathogenesis, development, and progression of various human pathologies, including multiple fibrosis and tumors. Due to the above fact, there is great interest in the abolition or modulation of EndMT as a new therapeutic approach for treating these disorders. Indeed, intensive research is currently being carried out focusing on the identification of new compounds, natural substances, and pharmacological agents intended to act as EndMT inhibitors and their use as a potential therapeutic agent in the treatment of various diseases in which EndMT is shown or suggested to play a role in their pathogenesis.
Targets postulated for these inhibitory effects are quite diverse. Still, a reasonably large group includes compounds with the ability to modulate the reorganization of the cytoskeleton as an essential cellular component directly responsible for the implementation of the EndMT process.
References
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 23, 5.
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411.
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42.
- Pacholczak, R.; Dropiński, J.; Walocha, J.; Musiał, J. Anti-cancer agents and endothelium. Oncol. Clin. Pract. 2018, 14, 249–256.
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010, 22, 631–637.
- Zeisberg, E.M.; Kalluri, R. Cellular mechanisms of tissue fibrosis, 1: Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C216–C225.
- Choi, S.H.; Hong, Z.Y.; Nam, J.K. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. 2015, 21, 3716–3726.
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287.
- Rieder, F.; Kessler, S.P.; West, G.A. Inflammation-induced endothelial-to-mesenchymal transition: A novel mechanism of intestinal fibrosis. Am. J. Pathol. 2011, 179, 2660–2673.
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961.
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128.
- Krizbai, I.A.; Gasparics, A.; Nagyoszi, P.; Fazakas, C.; Molnar, J.; Wilhelm, I.; Bencs, R.; Rosivall, L.; Sebe, A. Endothelial-mesenchymal transition of brain endothelial cells: Possible role during metastatic extravasation. PLoS ONE 2015, 10, e0119655.
- Wawro, M.E.; Chojnacka, K.; Wieczorek-Szukala, K.; Sobierajska, K.; Niewiarowska, J. Invasive colon cancer cells induce transdifferentiation of endothelium to cancer-associated fibroblasts through microtubules enriched in tubulin-beta3. Int. J. Mol. Sci. 2018, 20, 53.
- Fan, C.S.; Chen, W.S.; Chen, L.L.; Chen, C.C.; Hsu, Y.T.; Chua, K.V.; Wang, H.D.; Huang, T.S. Osteopontin-integrin engagement induces HIF-1α-TCF12-mediated endothelial-mesenchymal transition to exacerbate colorectal cancer. Oncotarget 2018, 9, 4998–5015.
- Yamada, N.O.; Heishima, K.; Akao, Y.; Senda, T. Extracellular vesicles containing microRNA-92a-3p facilitate partial endothelial-mesenchymal transition and angiogenesis in endothelial cells. Int. J. Mol. Sci. 2019, 20, 4406.
- Thannickal, V.J.; Zhou, Y.; Gaggar, A.; Duncan, S.R. Fibrosis: Ultimate and proximate causes. J. Clin. Investig. 2014, 124, 4673–4677.
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899.
- Bochaton-Piallat, M.L.; Gabbiani, G.; Hinz, B. The myofibroblast in wound healing and fibrosis: Answered and unanswered questions. F1000Research 2016, 26, 5, F1000 Faculty Rev-752.
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157.
- Pu, K.M.; Sava, P.; Gonzalez, A.L. Microvascular targets for anti-fibrotic therapeutics. Yale J. Biol. Med. 2013, 86, 537–554.
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529.
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999.
- Armstrong, E.J.; Bischoff, J. Heart valve development: Endothelial cell signaling and differentiation. Circ. Res. 2004, 95, 459–470.
- Mercado-Pimentel, M.E.; Runyan, R.B. Multiple transforming growth factor-beta isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs 2007, 185, 146–156.
- Pérez, L.; Muñoz-Durango, N.; Riedel, C.A.; Echeverría, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54.
- Sobierajska, K.; Ciszewski, W.M.; Sacewicz-Hofman, I.; Niewiarowska, J. Endothelial Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 71–86.
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011, 437, 515–520.
- Song, S.; Zhang, R.; Cao, W.; Fang, G.; Yu, Y.; Wan, Y.; Wang, C.; Li, Y.; Wang, Q. Foxm1 is a critical driver of TGF-β-induced EndMT in endothelial cells through Smad2/3 and binds to the Snail promoter. J. Cell. Physiol. 2019, 234, 9052–9064.
- Katagiri, T.; Tsukamoto, S.; Nakachi, Y.; Kuratani, M. Recent Topics in Fibrodysplasia Ossificans Progressiva. Endocrinol. Metab. (Seoul) 2018, 33, 331–338.
- Corada, M.; Nyqvist, D.; Orsenigo, F.; Caprini, A.; Giampietro, C.; Taketo, M.M.; Iruela-Arispe, M.L.; Adams, R.H.; Dejana, E. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev. Cell 2010, 18, 938–949.
- Chang, A.C.; Garside, V.C.; Fournier, M.; Smrz, J.; Vrljicak, P.; Umlandt, P.; Fuller, M.; Robertson, G.; Zhao, Y.; Tam, A.; et al. A Notch-dependent transcriptional hierarchy promotes mesenchymal transdifferentiation in the cardiac cushion. Dev. Dyn. 2014, 243, 894–905.
- Gasperini, P.; Espigol-Frigole, G.; McCormick, P.J.; Salvucci, O.; Maric, D.; Uldrick, T.S.; Polizzotto, M.N.; Yarchoan, R.; Tosato, G. Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through Notch-dependent signaling. Cancer Res. 2012, 72, 1157–1169.
- Lin, Q.-Q.; Zhao, J.; Zheng, C.-G.; Chun, J. Roles of Notch signaling pathway and endothelial-mesenchymal transition in vascular endothelial dysfunction and atherosclerosis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6485–6491.
- Fang, Y.; Chang, Z.; Xu, Z.; Hu, J.; Zhou, H.; Yu, S.; Wan, X. Osteoglycin silencing exerts inhibitory effects on myocardial fibrosis and epithelial/endothelial-mesenchymal transformation in a mouse model of myocarditis. Biofactors 2020, 46, 1018–1030.
- Jia, W.; Wang, Z.; Gao, C.; Wu, J.; Wu, Q. Trajectory modeling of endothelial-to-mesenchymal transition reveals galectin-3 as a mediator in pulmonary fibrosis. Cell Death Dis. 2021, 12, 327.
- Sabbineni, H.; Verma, A.; Somanath, P.R. Isoform-Specific Effects of Transforming Growth Factor-β on Endothelial to Mesenchymal Transition. J. Cell. Physiol. 2018, 233, 8418–8428.
- Giordo, R.; Ahmed, Y.M.A.; Allam, H.; Abusnana, S.; Pappalardo, L.; Nasrallah, G.K.; Mangoni, A.A.; Pintus, G. EndMT Regulation by Small RNAs in Diabetes-Associated Fibrotic Conditions: Potential Link with Oxidative Stress. Front. Cell Dev. Biol. 2021, 9, 683594.
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C.; et al. The role of PDGF-B/TGF-β1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell Signal. 2016, 28, 1489–1501.
- Thuan, D.T.B.; Zayed, H.; Eid, A.H.; Abou-Saleh, H.; Nasrallah, G.K.; Mangoni, A.A.; Pintus, G. A Potential Link Between Oxidative Stress and Endothelial-to-Mesenchymal Transition in Systemic Sclerosis. Front. Immunol. 2018, 9, 1985.
- Li, Z.; Chen, B.; Dong, W.; Kong, M.; Shao, Y.; Fan, Z.; Yu, L.; Wu, D.; Lu, J.; Guo, J.; et al. The Chromatin Remodeler Brg1 Integrates ROS Production and Endothelial-Mesenchymal Transition to Promote Liver Fibrosis in Mice. Front. Cell. Dev. Biol. 2019, 7, 245.
- Krenning, G.; Barauna, V.G.; Krieger, J.E.; Harmsen, M.C.; Moonen, J.R. Endothelial Plasticity: Shifting Phenotypes through Force Feedback. Stem Cells Int. 2016, 2016, 9762959.
- Balachandran, K.; Alford, P.W.; Wylie-Sears, J.; Goss, J.A.; Grosberg, A.; Bischoff, J.; Aikawa, E.; Levine, R.A.; Parker, K.K. Cyclic strain induces dual-mode endothelialmesenchymal transformation of the cardiac valve. Proc. Natl. Acad. Sci. USA 2011, 108, 19943–19948.
- Mai, J.; Hu, Q.; Xie, Y.; Su, S.; Qiu, Q.; Yuan, W.; Yang, Y.; Song, E.; Chen, Y.; Wang, J. Dyssynchronous pacing triggers endothelialmesenchymal transition through heterogeneity of mechanical stretch in a canine model. Circ. J. 2014, 79, 201–209.
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115.
- Moonen, J.R.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; van Kooten, T.G.; van Luyn, M.J.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 2015, 108, 377–386.
- Ma, X.; Zhao, D.; Yuan, P.; Li, J.; Yun, Y.; Cui, Y.; Zhang, T.; Ma, J.; Sun, L.; Ma, H.; et al. Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease. Acta Cardiol. Sin. 2020, 36, 183–194.
- Ma, J.; van der Zon, G.; Sanchez-Duffhues, G.; Ten Dijke, P. TGF-β-mediated Endothelial to Mesenchymal Transition (EndMT) and the Functional Assessment of EndMT Effectors using CRISPR/Cas9 Gene Editing. J. Vis. Exp. 2021, 26, 168.
- Man, S.; Sanchez Duffhues, G.; Ten Dijke, P.; Baker, D. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis 2019, 2, 3–13.
- Schliwa, M.; van Blerkom, J. Structural interaction of cytoskeletal components. J. Cell Biol. 1981, 90, 222–235.
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. How Cells Regulate Their Cytoskeletal Filaments. In Molecular Biology of the Cell, 4th ed.; Wilson, J., Hunt, T., Eds.; Garland Science: New York, NY, USA, 2002.
- Ramaekers, F.C.; Bosman, F.T. The cytoskeleton and disease. J. Pathol. 2004, 204, 351–354.
More
Information
Subjects:
Cell Biology; Oncology; Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
16 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No