 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christian Faul | + 1538 word(s) | 1538 | 2021-10-29 05:07:26 | | | |
| 2 | Catherine Yang | + 5 word(s) | 1543 | 2021-11-15 02:30:10 | | |
Video Upload Options
FGF23 is a bone-derived hormone that is essential for regulating vitamin D and phosphate homeostasis.
1. The Evolution and Biology of FGF23
Nearly 50 million years ago, life on earth was flourishing due to the re-population from terrestrial creatures, which progressed life beyond their aquatic habitats [1][2][3][4][5]. Throughout this period, an abundant amount of gene families, including the fibroblast growth factor (FGF) family, expanded in two phases which managed these new demands on all terrestrial systems, in comparison to their aquatic ancestors [6][7][8]. FGFs have been identified in both invertebrates and vertebrates. Prior to chordate evolution, the FGF gene family expanded from three to six genes via gene duplication, during their first phase of the metazoan linage [9]. During the early emergence and evolution of vertebrates, the FGF gene family underwent two large-scale gene duplications during their second phase [9]. This resulted in FGFs acquiring more significant roles in biological processes, such as in embryonic development, organogenesis and metabolic homeostasis [9][10][11].
The highly conserved FGF family consists of 22 members in mammals and is divided into three classifications: intracellular, paracrine and endocrine FGFs, based on their mechanisms of action [9][12]. Their conserved intrinsic core domain, which spans ~120 amino acids in length (~30–60% amino acid identity among all FGF members) promotes ligand binding to a distinct superfamily of receptor tyrosine kinases, known as fibroblast growth factor receptors (FGFRs) [6][9][13][14][15][16]. Intracellular FGFs are not secreted by their producing cells and act in an FGFR-independent manner, which enables them to mediate intracellular signaling events [9][17][18]. To date, their notable functions are regulating voltage-gated Na+ channels and the electrical excitability of neuronal cells [17][19][20][21]. Paracrine FGFs and endocrine FGFs (also called FGF19 subfamily) mediate their biological responses in an FGFR-dependent manner [6][9]. Unlike paracrine FGFs, which bind heparin or heparin sulfate proteoglycans (HPG) as a co-factor to facilitate FGFR activation and function as differentiation factors in development, endocrine FGFs have reduced affinity for HPG due to topological disparities in their heparin-binding region [9][22][23][24]. This permits the FGF19 subfamily to escape extracellular matrices and operate over long distances, functioning as circulating hormones [25][26][27]. As an alternative, endocrine FGFs employ αKlotho or βKlotho as their co-receptor to promote efficient FGF:FGFR binding [27].
FGF23 is a member of the FGF19 subfamily, which utilizes αKlotho to carry out its physiological functions, targeting the kidney to promote phosphate excretion and enabling the suppression of 1-25-dihydroxyvitamin D synthesis [9][27][28][29][30][31]. The development of this FGF23:αKlotho network is a ramification of vertebrate evolution, where primitive piscine ancestors such as ostracoderms acquired a boney endoskeleton [32]. In 2000, FGF23 was ultimately identified in the ventrolateral thalamic nucleus of mice, and its biological significance rapidly followed when a missense mutation of the Fgf23 gene in patients with autosomal dominant hypophosphatemic rickets (ADHR) was later identified [33][34]. Under physiologic conditions, FGF23 is predominantly produced by osteocytes as a 32 kDa glycoprotein in response to elevations in serum phosphate levels due to dietary loading or serum 1-25-dihydroxyvitamin D, where its N-terminal region shares homologies with other FGF family members and interacts with FGFRs, whereas its C-terminal portion binds αKlotho [30][35][36][37][38][39].
Prior to FGF23 secretion from bone, post-translational modifications allows this protein to circulate in the bloodstream as a full-length mature form (also called intact FGF23), which possess biological activity, or circulate as proteolytic cleaved fragments (Figure 1) [34][36][40][41]. This proteolytic cleavage event is executed by subtilisin-like pro-protein convertases, such as furin, which occurs at the consensus sequence Arg176-X-X-Arg179 and is not present in other FGF family members [34][40][42].
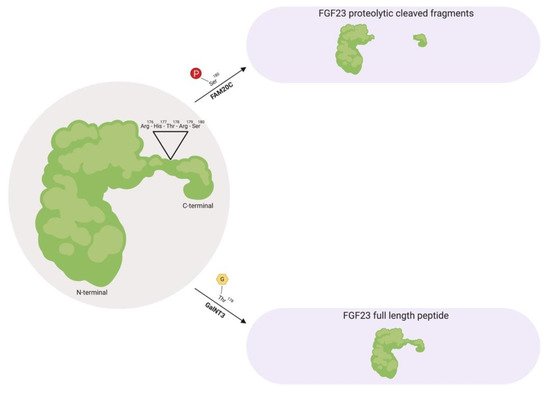
Figure 1. Fibroblast growth factor 23 (FGF23) regulation by post-translational modification. As a full-length biologically active protein in circulation, FGF23 is O-glycosylated by GalNT3 at several residues such as Thr178, which protects FGF23 from proteolytic cleavage by pro-protein convertases such as furin. Vice versa, as proteolytic cleaved fragments in circulation, FAM20C phosphorylates FGF23 at multiple amino acids, such as Ser180. This phosphorylation event impedes O-glycosylation by GalNT3 and allows furin to recognize its Arg176-His177-Thr178-Arg179-Ser180 consensus sequence in FGF23, thus leading to FGF23 cleavage and separation of the N-terminal and C-terminal fragments. GalNT3, polypeptide N-acetylgalactosaminyltransferase 3; FAM20C, secretory protein kinase family with sequence similarity-20 member C; Arg, arginine; His, histidine; Thr, threonine; Ser, serine.
As a circulating biologically active protein, FGF23 is O-glycosylated at several residues, such as Thr178 via polypeptide N-acetylgalactosaminyltransferase 3 (GalNT3), which protects FGF23 from proteolytic cleavage and helps support its intact structure [40][43][44][45]. The half-life of this biologically active form of FGF23 is ~45–60 minutes in humans, whereas in rodents, appears to be shorter with ~20–30 minutes in mice and ~5 minutes in rats [46][47][48].
Furthermore, as proteolytic cleaved fragments in circulation, FGF23 is phosphorylated at multiple amino acid residues, such as Ser180 via secretory protein kinase family with sequence similarity-20 member C (FAM20C), which impedes O-glycosylation at Thr178 by GalNT3 and results in its biologically inactive form by separating the respective FGFR and αKlotho binding regions [49][50]. A firm regulation of these post-translational modification and processing events are essential for maintaining proper homeostatic balances. Missense mutations in the FGF23 consensus sequence or in GalNT3 and/or FAM20C have been shown to be detrimental to physiologic processes, where interfering with FGF23 cleavage results in either elevated serum intact FGF23 levels that results in hypophosphatemia, or diminished serum intact FGF23 levels which results in hyperphosphatemia due to excessive proteolytic cleavage [34][43][51][52][53][54][55].
Regarding the metabolic rate of FGF23, renal extraction appears to play a critical role in its metabolism, yet only a minor contribution of FGF23 excretion via the kidney may contribute since FGF23 is not detectable in urine under physiological conditions [48]. However, in patients with acute kidney injury (AKI), FGF23 can be measured following urine analysis, where these elevations correspond with all-cause mortality [56]. Future investigations are needed in this context to help distinguish the origin of urinary FGF23.
2. FGF23 in Chronic Kidney Disease
The kidneys are part of the bone-kidney-intestinal axis [32]. The crucial role of this axis is to regulate mineral metabolism by altering tubular resorption of serum calcium and phosphate, with the assistance of key modulators FGF23, PTH and 1-25-dihydroxyvitamin D [57][58]. In an average Western diet, individuals ingest ~1200 mg/day of phosphate, yet a net weight of ~900 mg/day is absorbed into circulation [59][60][61]. The amount of phosphate absorption depends on its exogenous source and bioavailability [62][63]. Phosphate exists in two forms, organic and inorganic phosphate [57][63].
Organic phosphate is predominantly found in foods that are rich in protein (non-phytates) such as meat, fish and dairy products, which have a bioavailability of 40–80% [57][63][64][65][66]. By contrast, organic phosphate can also be obtained from plant-derived foods (phytates) such as cereal and nuts, but they have a bioavailability less than 40% [57][67][68][69]. This dissimilarity in phosphate absorption is attributed to monogastric mammals lacking the enzyme phytase, which is acquired to liberate phosphate [57][62][63][70]. To increase dietary absorption, phosphate additives are utilized to increase shelf life, alter texture and increase the flavor of food products [71][72][73][74]. These inorganic phosphate sources are passively absorbed in the intestine and have a bioavailability of nearly 100% [32][57][75][76]. With a wide range of foods containing additives such as polyphosphates and pyrophosphates, the relation of nephrotoxicity to phosphate toxicity (phosphotoxicity) has been the focus of many studies [32][63][77][78][79][80][81]. Clinical studies have shown that a single dose of 11.5 g of phosphate can accelerate de novo CKD over the course of several months [32].
To prevent phosphotoxicity, FGF23 levels rise and fall in correlation with the amount of dietary phosphate absorbed [82][83]. When an individual absorbs phosphate from foods that are high in its bioavailability, the bone upregulates FGF23 production to induce greater urinary excretion of phosphate and minimizing the efficiency of intestinal phosphate absorption by reducing serum 1-25-dihydroxyvitamin D levels [82]. In contrast, when an individual absorbs phosphate from foods that are low in its bioavailability, renal phosphate reabsorption is enhanced, as well as its absorption efficiency from the intestine by the actions of 1-25-dihydroxyvitamin D [82]. When renal damage occurs and kidney function declines, the alterations in bone and mineral homeostasis is inescapable. This event is termed chronic kidney disease-mineral bone disorder (CKD-MDB), which refers to renal dysfunction and altered levels of calcium, phosphate, PTH, 1-25-dihydroxyvitamin D and FGF23 [83][84].
During the early stages of CKD, the expression of the renal FGF23 co-receptor, αKlotho, decreases in response to kidney damage and progressively declines along with the loss of functional nephrons, promoting partial resistance to FGF23′s physiologic actions [82][85][86][87]. As a compensatory mechanism, FGF23 levels rise 1000-fold above normal values in an attempt to maintain a neutral phosphate balance [82][41][88]. This compensatory increase in FGF23 promotes the suppression of 1-25-dihydroxyvitamin D production, which in turn, promotes the elevation of PTH causing secondary hyperparathyroidism [89]. Throughout these hormonal alterations, serum phosphate levels gradually increase and by end-stage renal disease (ESRD), ultimately result in overt hyperphosphatemia due to renal resistance to FGF23′s actions on impaired kidneys [59][82][89].
These metabolic alterations primarily contribute to associated pathologies that are observed in CKD, such as immune dysfunction, systemic inflammation, anemia, vascular calcification, skeletal muscle dysfunction and cardiac hypertrophy, resulting in premature death [90][91][92][93][94][95][96]. Although FGF23 acts in a compensatory manner towards these elevating phosphate levels, clinical CKD studies have demonstrated powerful and dose-dependent associations between elevations in serum levels of FGF23 and CKD-associated pathologies, such as systemic inflammation, anemia and cardiovascular mortality, which is the leading cause of death across all stages of CKD [97][98][99][100][101]. FGF23 has also been shown to directly promote immune dysfunction, systemic inflammation and cardiac hypertrophy [102][103][104][105].
References
- Nam, K.; Lee, K.W.; Chung, O.; Yim, H.-S.; Cha, S.-S.; Lee, S.-W.; Jun, J.; Cho, Y.S.; Bhak, J.; de Magalhães, J.P.; et al. Analysis of the FGF gene family provides insights into aquatic adaptation in cetaceans. Sci. Rep. 2016, 7, 40233.
- Thewissen, J.G.M.; Cooper, L.N.; Clementz, M.T.; Bajpai, S.; Tiwari, B.N. Whales originated from aquatic artiodactyls in the Eocene epoch of India. Nature 2007, 450, 1190–1194.
- Thewissen, J.G.M.; Williams, E.M. The Early Radiations of Cetacea (Mammalia): Evolutionary Pattern and Developmental Correlations. Annu. Rev. Ecol. Syst. 2002, 33, 73–90.
- Masato, N.; Alejandro, R.; Okada, N. Phylogenetic relationships among cetartiodactyls based on insertions of short and long interspersed elements: Hippopotamuses are the closest extant relatives of whales. Proc. Natl. Acad. Sci. USA 1999, 96, 10261–10266.
- Gatsey, J.; OLeary, M. Deciphering whale origins with molecules and fossils. Trends Ecol. Evol. 2001, 16, 562–570.
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569.
- Sidow, A. Gen(om)e duplications in the evolution of early vertebrates. Curr. Opin. Genet. Dev. 1996, 6, 715–722.
- Horton, A.; Mahadevan, N. Phylogenetic Analyses Alone Are Insufficient to Determine Whether Genome Duplication(s) Occurred During Early Vertebrate Evolution. J. Exp. Zool. B Mol. Dev. Evol. 2003, 299, 41–53.
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130.
- Itoh, N. The Fgf Families in Humans, Mice, and Zebrafish: Their Evolutional Processes and Roles in Development, Metabolism, and Disease. Biol. Pharm. Bull. 2007, 30, 1819–1825.
- Fon Tacer, K.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research Resource: Comprehensive Expression Atlas of the Fibroblast Growth Factor System in Adult Mouse. Mol. Endocrinol. 2010, 24, 2050–2064.
- Itoh, N.; Ornitz, D.M. Functional evolutionary history of the mouse Fgf gene family. Dev. Dyn. 2007, 237, 18–27.
- Zhang, J.; Cousens, L.; Barr, P.; Sprang, S. Three-dimensional structure of human basic fibroblast growth factor a structural homolog of interleukin. Proc. Natl. Acad. Sci. 1991, 88, 3446–3450.
- Eriksson, A.E.; Cousens, L.; Mastthews, B. Three-dimensional structure of human basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA 1991, 88, 3441–3445.
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149.
- Turner, N.; Grose, R. Fibroblast growth factor signaling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129.
- Goldfarb, M.; Schoorlemmer, J.; Williams, A.; Diwakar, S.; Wang, Q.; Huang, X.; Giza, J.; Tchetchik, D.; Kelley, K.; Vega, A.; et al. Fibroblast Growth Factor Homologous Factors Control Neuronal Excitability through Modulation of Voltage-Gated Sodium Channels. Neuron 2007, 55, 449–463.
- Schoorlemmer, J.; Goldfarb, M. Fibroblast Growth Factor Homologous Factors and the Islet Brain-2 Scaffold Protein Regulate Activation of a Stress-activated Protein Kinase. J. Biol. Chem. 2002, 277, 49111–49119.
- Xiao, M.; Xu, L.; Laezza, F.; Yamada, K.; Feng, S.; Ornitz, D.M. Impaired hippocampal synaptic transmission and plasticity in mice lacking fibroblast growth factor 14. Mol. Cell. Neurosci. 2007, 34, 366–377.
- Shakkottai, V.G.; Xiao, M.; Xu, L.; Wong, M.; Nerbonne, J.M.; Ornitz, D.M.; Yamada, K.A. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol. Dis. 2009, 33, 81–88.
- Dover, K.; Solinas, S.; D’Angelo, E.; Goldfarb, M. Long-term inactivation particle for voltage-gated sodium channels. J. Physiol. 2010, 588, 3695–3711.
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, REVIEWS3005.
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular Insights into the Klotho-Dependent, Endocrine Mode of Action of Fibroblast Growth Factor 19 Subfamily Members. Mol. Cell. Biol. 2007, 27, 3417–3428.
- Harmer, N.J.; Pellegrini, L.; Chirgadze, D.; Fernandez-Recio, J.; Blundell, T.L. The Crystal Structure of Fibroblast Growth Factor (FGF) 19 Reveals Novel Features of the FGF Family and Offers a Structural Basis for Its Unusual Receptor Affinity. Biochemistry 2004, 43, 629–640.
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137.
- Angelin, B.; Larsson, T.E.; Rudling, M. Circulating Fibroblast Growth Factors as Metabolic Regulators—A Critical Appraisal. Cell Metab. 2012, 16, 693–705.
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signaling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180.
- Quarles, L.D. Endocrine functions of bone in mineral metabolism regulation. J. Clin. Investig. 2008, 118, 3820–3828.
- Erben, R.G.; Andrukhova, O. FGF23 regulation of renal tubular solute transport. Curr. Opin. Nephrol. Hypertens. 2015, 24, 450–456.
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a Potent Regulator of Vitamin D Metabolism and Phosphate Homeostasis. J. Bone Miner. Res. 2003, 19, 429–435.
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568.
- Hu, M.C.; Shiizaki, K.; Kuro-o, M.; Moe, O.W. Fibroblast Growth Factor 23 and Klotho: Physiology and Pathophysiology of an Endocrine Network of Mineral Metabolism. Annu. Rev. Physiol. 2013, 75, 503–533.
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a Novel Fibroblast Growth Factor, FGF-23, Preferentially Expressed in the Ventrolateral Thalamic Nucleus of the Brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498.
- White, K.E.; Carn, G.; Lorenz-Depiereux, B.; Benet-Pagès, A.; Strom, T.M.; Econs, M.J. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001, 60, 2079–2086.
- Shimada, T.; Yamazaki, Y.; Takahashi, M.; Hasegawa, H.; Urakawa, I.; Oshima, T.; Ono, K.; Kakitani, M.; Tomizuka, K.; Fujita, T.; et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am. J. Ren. Physiol. 2005, 289, F1088–F1095.
- Martin, A.; David, V.; Quarles, L.D. Regulation and Function of the FGF23/Klotho Endocrine Pathways. Physiol. Rev. 2012, 92, 131–155.
- Yamazaki, Y.; Tamada, T.; Kasai, N.; Urakawa, I.; Aono, Y.; Hasegawa, H.; Fujita, T.; Kuroki, R.; Yamashita, T.; Fukumoto, S.; et al. Anti-FGF23 Neutralizing Antibodies Show the Physiological Role and Structural Features of FGF23. J. Bone Miner. Res. 2008, 23, 1509–1518.
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412.
- Xu, Y.; Sun, Z. Molecular Basis of Klotho: From Gene to Function in Aging. Endocr. Rev. 2015, 36, 174–193.
- Shimada, T.; Muto, T.; Urakawa, I. Mutant FGF-23 responsible for autosomal dominant hypophsophatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinolgy 2002, 143, 3179–3182.
- Yamazaki, Y.; Okazaki, R.; Hasegawa, Y.; Satoh, K.; Tajima, T.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Yamashita, T.; Fukumoto, S. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J. Clin. Endocrinol. Metab. 2002, 87, 4957–4960.
- Benet-Pagès, A.; Lorenz-Depiereux, B.; Zischka, H.; White, K.E.; Econs, M.J.; Strom, T.M. FGF23 is processed by proprotein convertases but not by PHEX. Bone 2004, 35, 455–462.
- Frishberg, Y.; Ito, N.; Rinat, C.; Yamazaki, Y.; Feinstein, S.; Urakawa, I.; Navon-Elkan, P.; Becker-Cohen, R.; Yamashita, T.; Araya, K.; et al. Hyperostosis-Hyperphosphatemia Syndrome: A Congenital Disorder of O-Glycosylation Associated with Augmented Processing of Fibroblast Growth Factor 23. J. Bone Miner. Res. 2006, 22, 235–242.
- Bergwitz, C.; Banerjee, S.; Abu-Zahra, H.; Kaji, H.; Miyauchi, A.; Sugimoto, T.; Jüppner, H. Defective O-Glycosylation due to a Novel Homozygous S129P Mutation Is Associated with Lack of Fibroblast Growth Factor 23 Secretion and Tumoral Calcinosis. J. Clin. Endocrinol. Metab. 2009, 94, 4267–4274.
- Kato, K.; Jeanneau, C.; Tarp, M. Polypeptide GalNAc-transferase T3 and Familial Tumoral Calcinosis. J. Biol. Chem. 2006, 281, 18370–18377.
- Khosravi, A.; Cutler, C.M.; Kelly, M.H.; Chang, R.; Royal, R.E.; Sherry, R.M.; Wodajo, F.M.; Fedarko, N.S.; Collins, M.T. Determination of the Elimination Half-Life of Fibroblast Growth Factor-23. J. Clin. Endocrinol. Metab. 2007, 92, 2374–2377.
- Christov, M.; Waikar, S.S.; Pereira, R.C.; Havasi, A.; Leaf, D.E.; Goltzman, D.; Pajevic, P.D.; Wolf, M.; Jüppner, H. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int. 2013, 84, 776–785.
- Mace, M.L.; Gravesen, E.; Hofman-Bang, J.; Olgaard, K.; Lewin, E. Key role of the kidney in the regulation of fibroblast growth factor 23. Kidney Int. 2015, 88, 1304–1313.
- Lindberg, I.; Pang, H.W.; Stains, J.P.; Clark, D.; Yang, A.J.; Bonewald, L.; Li, K.Z. FGF23 is endogenously phosphorylated in bone cells. J. Bone Miner. Res. 2015, 30, 449–454.
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Nidumanda Appaiah, H.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525.
- ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348.
- Wang, X.; Wang, S.; Li, C.; Gao, T.; Liu, Y.; Rangiani, A.; Sun, Y.; Hao, J.; George, A.; Lu, Y.; et al. Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice. PLoS Genet. 2012, 8, e1002708.
- Topaz, O.; Shurman, D.L.; Bergman, R.; Indelman, M.; Ratajczak, P.; Mizrachi, M.; Khamaysi, Z.; Behar, D.; Petronius, D.; Friedman, V.; et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat. Genet. 2004, 36, 579–581.
- Larsson, T.; Davis, S.I.; Garringer, H.J.; Mooney, S.D.; Draman, M.S.; Cullen, M.J.; White, K.E. Fibroblast Growth Factor-23 Mutants Causing Familial Tumoral Calcinosis Are Differentially Processed. Endocrinology 2005, 146, 3883–3891.
- Ichikawa, S.; Sorenson, A.H.; Austin, A.M.; Mackenzie, D.S.; Fritz, T.A.; Moh, A.; Hui, S.L.; Econs, M.J. Ablation of the GalnT3 Gene Leads to Low-Circulating Intact Fibroblast Growth Factor 23 (Fgf23) Concentrations and Hyperphosphatemia Despite Increased Fgf23 Expression. Endocrinology 2009, 150, 2543–2550.
- Leaf, D.E.; Jacob, K.A.; Srivastava, A.; Chen, M.E.; Christov, M.; Jüppner, H.; Sabbisetti, V.S.; Martin, A.; Wolf, M.; Waikar, S.S. Fibroblast Growth Factor 23 Levels Associate with AKI and Death in Critical Illness. J. Am. Soc. Nephrol. 2017, 28, 1877–1885.
- Vervloet, M.G.; Sezer, S.; Massy, Z.A.; Johansson, L.; Cozzolino, M.; Fouque, D. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 2016, 13, 27–38.
- Naveh-Many, T.; Silver, J. The Pas de Trois of Vitamin D, FGF23, and PTH. J. Am. Soc. Nephrol. 2017, 28, 393–395.
- Hruska, K.A.; Mathew, S.; Lund, R.; Qiu, P.; Pratt, R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008, 74, 148–157.
- Komaba, H.; Fukagawa, M. Phosphate—A poison for humans? Kidney Int. 2016, 90, 753–763.
- Vervloet, M.G.; van Ballegooijen, A.J. Prevention and treatment of hyperphosphatemia in chronic kidney disease. Kidney Int. 2018, 93, 1060–1072.
- Fukagawa, M.; Komaba, H.; Miyamoto, K.-I. Source matters: From phosphorus load to bioavailability. Clin. J. Am. Soc. Nephrol. 2011, 6, 239–240.
- Kalantar-Zadeh, K.; Gutekunst, L.; Mehrotra, R.; Kovesdy, C.P.; Bross, R.; Shinaberger, C.S.; Noori, N.; Hirschberg, R.; Benner, D.; Nissenson, A.R.; et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 519–530.
- Boaz, M.; Smetana, S. Regression Equation Predicts Dietary Phosphorus Intake from Estimate of Dietary Protein Intake. J. Am. Diet. Assoc. 1996, 96, 1268–1270.
- Kayne, L.H.; D’Argenio, D.Z.; Meyer, J.H.; Hu, M.S.; Jamgotchian, N.; Lee, D.B. Analysis of segmental phosphate absorption in intact rats. A compartmental analysis approach. J. Clin. Investig. 1993, 91, 915–922.
- Uribarri, J. Phosphorus Metabolism and Management in Chronic Kidney Disease: Phosphorus Homeostasis in Normal Health and in Chronic Kidney Disease Patients with Special Emphasis on Dietary Phosphorus Intake. Semin. Dial. 2007, 20, 295–301.
- Schlemmer, U.; Frølich, W. Phytate in foods and significance for humans: Food sources, intake, processing, bioavailability, protective role and analysis. Mol. Nutr. 2009, 53, S330–S375.
- Sandberg, A.S.; Andersson, H.; Kivistö, B.; Sandström, B. Extrusion cooking of a high-fiber cereal product. Br. J. Nutr. 1986, 55, 245–254.
- Waldroup, P.W.; Kersey, J.H.; Saleh, E.A. Nonphytate Phosphorus Requirement and Phosphorus Excretion of Broiler Chicks Fed Diets Composed of Normal or High Available Phosphate Corn with and Without Microbial Phytase. Poult. Sci. 2000, 79, 1451–1459.
- Lei, X.G.; Porres, J.M. Phytase enzymology, applications, and biotechnology. Biotechnol. Lett. 2003, 25, 1787–1794.
- Ritz, E.; Hahn, K.; Ketteler, M.; Kuhlmann, M.K.; Mann, J. Phosphate Additives in Food. Deutsch. Aerztebl. Int. 2012, 109, 49–55.
- Sullivan, C.; Sayre, S.S.; Leon, J.B.; Machekano, R.; Love, T.E.; Porter, D.; Marbury, M.; Sehgal, A.R. Effect of Food Additives on Hyperphosphatemia Among Patients with End-stage Renal Disease. JAMA 2009, 301, 629–635
- DSc, O.B.; RD, C.D.; VS, D.G.; PhD, A.C.M. Extra-Phosphate Load From Food Additives in Commonly Eaten Foods: A Real and Insidious Danger for Renal Patients. J. Ren. Nutr. 2011, 21, 303–308.
- Karalis, M.; Murphy-Gutekunst, L. Enhanced Foods: Hidden Phosphorus and Sodium in Foods Commonly Eaten. J. Ren. Nutr. 2006, 16, 79–81.
- Kemi, V.E.; Rita, H.J.; Kärkkäinen, M.U.; Viljakainen, H.T.; Laaksonen, M.M.; Outila, T.A.; Lamberg-Allardt, C.J. Habitual high phosphorus intakes and foods with phosphate additives negatively affect serum parathyroid hormone concentration: A cross-sectional study on healthy premenopausal women. Public Health Nutr. 2009, 12, 1885–1892.
- Uribarri, J. Hidden Sources of Phosphorus in the Typical American Diet: Does it Matter in Nephrology? Semin. Dial. 2003, 16, 186–188.
- Calvo, M. Dietary Considerations to Prevent Loss of Bone and Renal Function. Nutrition 2000, 16, 564–566.
- León, J.B.; Sullivan, C.M.; Sehgal, A.R. The Prevalence of Phosphorus-Containing Food Additives in Top-Selling Foods in Grocery Stores. J. Ren. Nutr. 2013, 23, 265–270.
- Moser, M.; White, K.; Henry, B.; Oh, S.; Miller, E.R.; Anderson, C.A.; Benjamin, J.; Charleston, J.; Appel, L.J.; Chang, A.R. Phosphorus Content of Popular Beverages. Am. J. Kidney Dis. 2015, 65, 969–971.
- Bell, R.R.; Draper, H.H.; Tzeng, D.Y.M.; Shin, H.K.; Schmidt, G.R. Physiological Responses of Human Adults to Foods Containing Phosphate Additives. J. Nutr. 1977, 107, 42–50.
- Shafey, T.M.; McDonald, M.W.; Pym, R.A.E. Effects of dietary calcium, available phosphorus and vitamin D on growth rate, food utilization, plasma and bone constituents and calcium and phosphorus retention of commercial broiler strains. Br. Poult. Sci. 1990, 31, 587–602.
- Wolf, M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012, 82, 737–747.
- Jüppner, H. Phosphate and FGF-23. Kidney Int. 2011, 79, S24–S27.
- Graciolli, F.G.; Neves, K.R.; Barreto, F.; Barreto, D.V.; Reis, dos, L.M.; Canziani, M.E.; Sabbagh, Y.; Carvalho, A.B.; Jorgetti, V.; Elias, R.M.; et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017, 91, 1436–1446.
- Drew, D.A.; Katz, R.; Kritchevsky, S.; Ix, J.; Shlipak, M.; Gutiérrez, O.M.; Newman, A.; Hoofnagle, A.; Fried, L.; Semba, R.D.; et al. Association between Soluble Klotho and Change in Kidney Function: The Health Aging and Body Composition Study. J. Am. Soc. Nephrol. 2017, 28, 1859–1866.
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Klotho and Chronic Kidney Disease. Contrib. Nephrol. 2013, 180, 47–63.
- Di Zou; Wu, W.; He, Y.; Ma, S.; Gao, J. The role of klotho in chronic kidney disease. BMC Nephrol. 2018, 19, 285.
- Isakova, T.; Gutiérrez, O.M.; Wolf, M. A blueprint for randomized trials targeting phosphorus metabolism in chronic kidney disease. Kidney Int. 2009, 76, 705–716.
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378.
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaeus, A.; Stenvinkel, P.; Lindholm, B. Aspects of Immune Dysfunction in End-stage Renal Disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1526–1533.
- Meuwese, C.L.; Stevinkel, P.; Carrero, J.J. Monitoring of inflammation in patients on dialysis: Forewarned is forearmed. Nat. Rev. Nephrol. 2011, 7, 166–176.
- Fishbane, S.; Spinowitz, B. Update on Anemia in ESRD and Earlier Stages of CKD: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 71, 423–435.
- Moe, S.M.; Chen, N.X. Mechanisms of Vascular Calcification in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2008, 19, 213–216.
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516.
- Mathew, J.; Katz, R.; St Sutton, M.J.; Dixit, S.; Gerstenfeld, E.P.; Ghio, S.; Gold, M.R.; Linde, C.; Shlipak, M.G.; Deo, R. Chronic kidney disease and cardiac remodeling in patients with mild heart failure: Results from the REsynchronization reVErses Remodeling in Systolic Left vEntricular Dysfunction (REVERSE) study. Eur. J. Heart Fail. 2014, 14, 1420–1428.
- Tonelli, M. Chronic Kidney Disease and Mortality Risk: A Systematic Review. J. Am. Soc. Nephrol. 2006, 17, 2034–2047.
- Gutiérrez, O.M.; Mannstadt, M. Fibroblast Growth Factor 23 and Mortality among Patients Undergoing Hemodialysis. N. Engl. J. Med. 2008, 359, 584–592.
- Isakova, T.; Wolf, M. Fibroblast Growth Factor 23 and Risks of Mortality and End-Stage Renal Disease in Patients with Chronic Kidney Disease. JAMA 2011, 305, 2432–2439.
- Kendrick, J.; Cheung, A.K.; Kaufman, J.S.; Greene, T.; Roberts, W.L.; Smits, G.; Chonchol, M.; HOST Investigators. FGF-23 Associates with Death, Cardiovascular Events, and Initiation of Chronic Dialysis. J. Am. Soc. Nephrol. 2011, 22, 1913–1922.
- Mendoza, J.M.; Isakova, T.; Cai, X.; Bayes, L.Y.; Faul, C.; Scialla, J.J.; Lash, J.P.; Chen, J.; He, J.; Navaneethan, S.; et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2016, 91, 711–719.
- Mehta, R.; Cai, X.; Hodakowski, A.; Lee, J.; Leonard, M.; Ricardo, A.; Chen, J.; Hamm, L.; Sondheimer, J.; Dobre, M.; et al. Fibroblast Growth Factor 23 and Anemia in the Chronic Renal Insufficiency Cohort Study. Clin. J. Am. Soc. Nephrol. 2017, 12, 1795–1803.
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032.
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996.
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974.
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408.

