 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Erika Di Zazzo | + 2959 word(s) | 2959 | 2021-11-09 11:10:45 | | | |
| 2 | Beatrix Zheng | + 256 word(s) | 3215 | 2021-11-11 04:11:24 | | | | |
| 3 | Beatrix Zheng | Meta information modification | 3215 | 2021-11-12 07:35:42 | | |
Video Upload Options
PRDM12 is a member of the PRDI-BF1 (positive regulatory domain I-binding factor 1) homologous domain (PRDM)-containing protein family, a subfamily of Kruppel-like zinc finger proteins, controlling key processes in the development of cancer. PRDM12 is expressed in a spatio-temporal manner in neuronal systems where it exerts multiple functions. PRDM12 is essential for the neurogenesis initiation and activation of a cascade of downstream pro-neuronal transcription factors in the nociceptive lineage.
1. PRDM12 is a member of PRDM gene family
2. Established PRDM12 Functions: Neurogenesis
In P19 embryonal carcinoma cells, an in in vitro mouse model systems for neurogenesis, retinoic acid (RA) that prompted neural differentiation into neurons and glial cells, induced Prdm12 expression, possibly through the regulation of a putative RA receptor (RAR)-beta response element. Additionally, Prdm12 overexpression impaired P19 cell proliferation and increased the percentage of cells in the G1 phase accompanied by p27 upregulation. Furthermore, both the PR domain and zinc finger domains were required for the anti-proliferative activity of PRDM12. In contrast, Prdm12 knockdown and Prdm12 mutants resulted in an increased number of cells in a suspension culture of RA-induced neural differentiation [11]. Altogether, these results suggested that Prdm12 was induced by the RA signaling and might control neural differentiation during development through p27 expression level regulation.
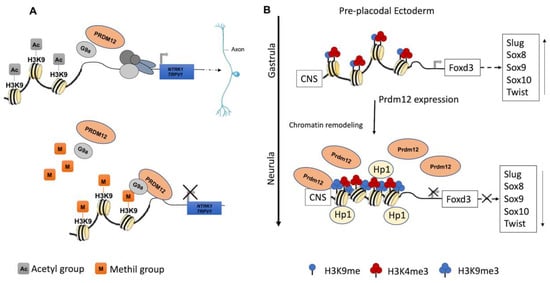
During the early neurula stage of Xenopus embryos, prdm12 expression was also revealed in the lateral pre-placodal ectoderm after the late gastrula stage (st. 13), where it was regulated by both BMP and Wnt signaling ( Figure 1 B) [12]. Several gain- and loss-of-function experiments were approached to clarify the role of Prdm12 in early Xenopus development. prdm12 overexpression through mRNA injection inhibited the expression of neural crest markers (Foxd3, Slug, Sox8, -9, -10 and Twist) via H3K9 trimethylation (H3K9me3) ( Figure 2 B). Otherwise, prdm12 knockdown through an antisense morpholino oligomer (MO) inhibited the expression of presumptive trigeminal placode markers and expanded the neural crest region through a H3K9me3 level decrease in the Foxd3 gene promoter ( Figure 2 B). Notably, the histone demethylase, Kdm4a, inhibited the expression of presumptive trigeminal placode markers producing a similar effect of prdm12 knockdown. Accordingly, ChIP-qPCR analyses revealed that the expression of H3K9me3 on the Foxd3, Slug, and Sox8 promoters was inhibited by Kdm4a overexpression. Altogether, the mutual relationship between Prdm12 and Kdm4a indicated that the modification of the H3K9 methylation levels on the neural crest gene promoters by these two proteins would determine a demarcation line between the pre-placodal ectoderm and the neural crest region [12]. Interestingly, a recent analysis, performed to screen for zic1 targets in the midbrain region of Xenopus , revealed that prdm12 was a downstream target of zic1 [13]. Zic1 is a highly conserved zinc finger transcription factor playing a critical role in the establishment of the nervous system; it is expressed on the lateral edge of the neural plate and in the dorsal neural tube [13]. Here, prdm12 was expressed in the caudal forebrain, midbrain and hindbrain. Moreover, during embryonic development, zic1 and prdm12 were co-expressed in the same cell, with Zic1 controlling the expression of prdm12 mediated by Wnt signaling during brain cell differentiation. Additionally, gain- and loss-of-function experiments revealed that prdm12 was both necessary and sufficient to promote midbrain formation in the embryo [13].
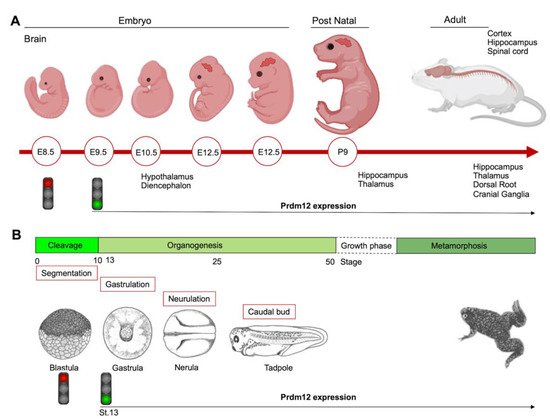
In addition to the central nervous system, prdm12 expression was also detected in the peripheral nervous system. The V1 interneurons are a class of inhibitory glycinergic neurons playing a conserved role in vertebrate locomotion; they originate from the spinal cord p1 domain and are characterized by the expression of Engrailed-1 (En1/Eng1). prdm12b , the zebrafish prdm12 homolog, was expressed in the p1 domain of the neural tube at least partially in response to Sonic Hedgehog (Shh) signaling. Interestingly, prdm12b disruption led to the inappropriate dorsoventral patterning of the neural tube, depletion of the V1 interneurons and an impaired escape response in zebrafish. These data suggest that prdm12b is a key component of the genetic program required for motor circuit formation [14]. Likewise, in the frog embryos, prdm12 was selectively expressed in p1 progenitors of the hindbrain and spinal cord; this restricted expression profile was also observed in the neural tube of chick embryos and in the ventral nerve cord of the larvae of the basal chordate amphioxus. Moreover, in frog, chicken and mice, Prdm12 expression in the p1 domain progenitors of the caudal neural tube was dependent on RA signaling and Pax6 and it was repressed by Dbx1 and Nkx6-1/2 expressed in the adjacent p0 and p2 domains [15]. Functional studies in Xenopus and the genome-wide identification of molecular targets by RNA-seq and ChIP-Seq, revealed that the vertebrate Prdm12 acted as a general determinant of V1 cell fate, at least in part, by directly repressing Dbx1 and Nkx6 genes. Both the PR and zinc-finger domains of Prdm12 were required to exert this function; specifically, Prdm12 may act as a G9a-dependent repressor to induce En1. However, this activity was not found in the amphioxus, and differences in the C-terminal region of the protein, including the zinc-finger domains, may account for the differential functions of the amphioxus and vertebrate proteins. Overall, these findings indicated that Prdm12 could promote V1 interneurons through cross-repressive interactions with Dbx1 and Nkx6 genes. Interestingly, this function could be acquired after the split between the vertebrate and cephalochordate lineages [15]. Recently, the analysis of CRISPR/Cas9 prdm12 mutants, recapitulating the phenotypes observed by MO-based approaches, has demonstrated that prdm12b acts as transcriptional repressor in zebrafish, and that it can interact with both EHMT2/ G9a and Bhlhe22, a member of the basic Helix-Loop-Helix (bHLH) family, through its zinc-finger domain. However, bhlhe22 function is not required for eng1b expression in vivo, suggesting that other bhlh genes could be involved during embryogenesis. This study also suggested that prdm12b is not only required to repress non-p1 fates, but also to promote p1 fates [16]. Additionally, a study in a mouse model revealed strong evidence that Dbx1 and Prdm12 expression was inhibited by both Pax3 and Pax7, two highly related transcription factors controlling the spatial organization of spinal differentiation [17]. Notably, another member of the Prdm family, PRDM13, was recently shown to be required for the restriction of Prdm12 expression to the ventral neural tube during mouse embryogenesis [18]. In mouse Prdm13 mutants, Prdm12 was aberrantly expressed in the dorsal region, altering the identity of these neurons. Mechanistically, PRDM13 interacted with the genomic regions, overlapping those bound by neural bHLH factors and functions, by limiting the ability of these bHLH factors to activate enhancer-driven reporters. Specifically, PRDM13 repressed Prdm12 in the dorsal neural tube via the inhibition of NEUROG1 and NEUROG2, which were likely to activate mouse Prdm12 transcription through one enhancer localized more than 25 kb upstream of the ATG starting site [18].
PRDM12 could also function in the vagal sensory nervous system, to maintain visceral homeostasis. Indeed, transcriptome profiling performed to reveal differentially expressed genes between nodose and jugular C-fiber neurons detected Prdm12 as preferentially expressed in mouse jugular vagal neurons [19].
3. Established PRDM12 Functions: Pain Perception
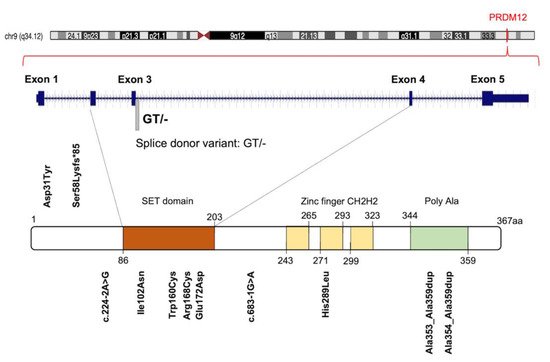
Initially, 10 different homozygous mutations in PRDM12 were identified in subjects from 11 families with a congenital insensitivity to pain (CIP), a type of hereditary sensory and autonomic neuropathy (HSAN), which is a clinically and genetically heterogeneous group of inherited neuropathies predominantly affecting peripheral sensory and autonomic neurons [20]. Most of the variants were missense mutations, despite the revelation of a splice-site mutation, a frame-shift mutation and an 18-alanine-repeat mutation (general population contains a maximum of 14-alanine in this polymorphic site; Figure 3 and Table S1 ). Heterozygote carriers were asymptomatic with a normal pain perception. To determine whether these mutations could cause developmental defects in the sensory neurons, committed to becoming nociceptors, the expression of PRDM12 during embryogenesis and the differentiation in various in vivo models (mouse, Xenopus and human iPSC derived sensory neurons) was explored. Prdm12 was expressed in nociceptors and their progenitors and participated in the development of sensory neurons [20]. Moreover, the CIP-associated PRDM12 mutations impaired the histone-methylation capacity [20]. In an independent study, Prdm12 was also investigated as a key regulator of sensory neuronal specification in Xenopus [20]. In this case, the modeling analysis of human PRDM12 mutations causing HSAN revealed a remarkable conservation of the mutated residues during evolution. As shown by RNAseq analyses, the expression of wild-type human PRDM12 in Xenopus induced the expression of several sensory neuronal markers including Islet1 and Tlx3; in contrast, embryos treated with PRDM12 MO or PRDM12 mutants displayed reduced levels of these markers [21]. In Drosophila , the Hamlet gene was identified as the functional PRDM12 homolog that controls nociceptive behavior in sensory neurons. Interestingly, the ectopic expression of human PRDM12 mutants in Drosophila nociceptor neurons impaired pain perception, thus supporting the idea that PRDM12 was an evolutionary, conserved, master regulator of sensory neuronal specification that played a critical role in pain perception [21]. In addition to that, RNAseq analyses of human patient fibroblasts with PRDM12 mutations disclosed the possible downstream target genes. Among them, the gene-encoding, thyrotropin-releasing, hormone-degrading enzyme ( TRHDE ) was revealed; its substrate, TRH, was previously found to affect pain in human and rodents. TRHDE knockdown in Drosophila sensory neurons resulted in an altered cellular morphology and impaired nociception. These findings also added to our knowledge that novel molecules and pathways controlled evolutionary, conserved nociception [21].
The upstream genes and signals controlling PRDM12 expression in developing sensory ganglia still remain to be addressed. It could be speculated that PRDM12, essential for TrkA initiation, is also a target of NGF-TrkA signaling, considering that, in adult mice and humans, NGF signaling induces nociceptor sensitization leading to chronic pain states [22] and PRDM12 is highly expressed in mature nociceptors. Interestingly, PRDM12 expression increased significantly (by 1000 times) when skin-derived precursor cells (a subtype of neural crest stem cells that persist in certain adult tissues such as the skin) were induced in vitro to differentiate into sensory neurons by several molecules, including NGF, which prompted the upregulation of neurogenins [23].
Moreover, both other partners of PRDM12 constituting the transcriptional complex, which epigenetically regulated gene expression in developing nociceptors, as well as the PRDM12 transcriptional targets need to be identified. Overall, these studies suggest that pharmacotherapies targeting this pathway, or the epigenetic mechanisms controlled by PRDM12, and could be a promising strategy in the treatment of chronic pain conditions.
The supplementary understandings of the identity of the potential PRDM12 interactors and the transcriptional, and epigenomic changes in the sensory neuron progenitors upon PRDM12 manipulation, will be relevant in understanding the PRDM12 gene regulation during the generation of the nociceptive lineage. The further comprehension of the involved molecular mechanisms will provide key insights into how sensory neuron diversity is generated and may provide genetic tools to induce a desired neuronal lineage in stem cell engineering.
4. Exploring Novel PRDM12 Functions: Cancer
5. Exploring Novel PRDM12 Functions: Cell Metabolism
References
- Casamassimi, A.; Rienzo, M.; Di Zazzo, E.; Sorrentino, A.; Fiore, D.; Proto, M.C.; Moncharmont, B.; Gazzerro, P.; Bifulco, M.; Abbondanza, C. Multifaceted Role of PRDM Proteins in Human Cancer. Int. J. Mol. Sci. 2020, 21, 2648.
- Di Zazzo, E.; De Rosa, C.; Abbondanza, C.; Moncharmont, B. PRDM Proteins: Molecular Mechanisms in Signal Transduction and Transcriptional Regulation. Biology 2013, 2, 107–141.
- Hayashi, K.; Yoshida, K.; Matsui, Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 2005, 438, 374–378.
- Eram, M.S.; Bustos, S.P.; Fernandes, E.L.; Siarheyeva, A.; Senisterra, G.; Hajian, T.; Chau, I.; Duan, S.; Wu, H.; Dombrovski, L.; et al. Trimethylation of Histone H3 Lysine 36 by Human Methyltransferase PRDM9 Protein. J. Biol. Chem. 2014, 289, 12177–12188.
- Pinheiro, I.; Margueron, R.; Shukeir, N.; Eisold, M.; Fritzsch, C.; Richter, F.M.; Mittler, G.; Genoud, C.; Goyama, S.; Kurokawa, M.; et al. Prdm3 and Prdm16 are H3K9me1 Methyltransferases Required for Mammalian Heterochromatin Integrity. Cell 2012, 150, 948–960.
- Hohenauer, T.; Moore, A.W. The Prdm family: Expanding roles in stem cells and development. Development 2012, 139, 2267–2282.
- Sorrentino, A.; Federico, A.; Rienzo, M.; Gazzerro, P.; Bifulco, M.; Ciccodicola, A.; Casamassimi, A.; Abbondanza, C. PR/SET Domain Family and Cancer: Novel Insights from the Cancer Genome Atlas. Int. J. Mol. Sci. 2018, 19, 3250.
- Di Tullio, F.; Schwarz, M.; Zorgati, H.; Mzoughi, S.; Guccione, E. The duality of PRDM proteins: Epigenetic and structural perspectives. FEBS J. 2021.
- Rienzo, M.; Sorrentino, A.; Di Zazzo, E.; Di Donato, M.; Carafa, V.; Marino, M.M.; De Rosa, C.; Gazzerro, P.; Castoria, G.; Altucci, L.; et al. Searching for a Putative Mechanism of RIZ2 Tumor-Promoting Function in Cancer Models. Front. Oncol. 2021, 10, 583533.
- Mzoughi, S.; Tan, Y.X.; Low, D.; Guccione, E. The role of PRDMs in cancer: One family, two sides. Curr. Opin. Genet. Dev. 2016, 36, 83–91.
- Yang, C.-M.; Shinkai, Y. Prdm12 Is Induced by Retinoic Acid and Exhibits Anti-proliferative Properties through the Cell Cycle Modulation of P19 Embryonic Carcinoma Cells. Cell Struct. Funct. 2013, 38, 197–206.
- Matsukawa, S.; Miwata, K.; Asashima, M.; Michiue, T. The requirement of histone modification by PRDM12 and Kdm4a for the development of pre-placodal ectoderm and neural crest in Xenopus. Dev. Biol. 2015, 399, 164–176.
- Rahman, M.; Kim, I.; Ahn, D.; Tae, H.; Park, B. PR domaincontaining protein 12 (prdm12) is a downstream target of the transcription factor zic1 during cellular differentiation in the central nervous system: PR domain containing protein is the right form. Int. J. Dev. Neurosci. 2020, 80, 528–537.
- Zannino, D.A.; Downes, G.B.; Sagerström, C.G. prdm12b specifies the p1 progenitor domain and reveals a role for V1 interneurons in swim movements. Dev. Biol. 2014, 390, 247–260.
- Thélie, A.; Desiderio, S.; Hanotel, J.; Quigley, I.; Van Driessche, B.; Rodari, A.; Borromeo, M.D.; Kricha, S.; Lahaye, F.; Croce, J.; et al. Prdm12 specifies V1 interneurons through cross-repressive interactions with Dbx1 and Nkx6 genes in Xenopus. Development 2015, 142, 3416–3428.
- Yildiz, O.; Downes, G.B.; Sagerström, C.G. Zebrafish prdm12b acts independently of nkx6.1 repression to promote eng1b expression in the neural tube p1 domain. Neural Dev. 2019, 14, 5.
- Gard, C.; Curto, G.G.; Frarma, Y.E.-M.; Chollet, E.; Duval, N.; Auzié, V.; Auradé, F.; Vigier, L.; Relaix, F.; Pierani, A.; et al. Pax3- and Pax7-mediated Dbx1 regulation orchestrates the patterning of intermediate spinal interneurons. Dev. Biol. 2017, 432, 24–33.
- Mona, B.; Uruena, A.; Kollipara, R.K.; Ackerman, S.L.; Borromeo, M.D.; Chang, J.C.; Johnson, J.E. Repression by PRDM13 is critical for generating precision in neuronal identity. eLife 2017, 6, e25787.
- Wang, J.; Kollarik, M.; Ru, F.; Sun, H.; McNeil, B.; Dong, X.; Stephens, G.; Korolevich, S.; Brohawn, P.; Kolbeck, R.; et al. Distinct and common expression of receptors for inflammatory mediators in vagal nodose versus jugular capsaicin-sensitive/TRPV1-positive neurons detected by low input RNA sequencing. PLoS ONE 2017, 12, e0185985.
- Chen, Y.-C.; Auer-Grumbach, M.; Matsukawa, S.; Zitzelsberger, M.; Themistocleous, A.; Strom, T.M.; Samara, C.; Moore, A.W.; Cho, L.T.-Y.; Young, G.T.; et al. Transcriptional regulator PRDM12 is essential for human pain perception. Nat. Genet. 2015, 47, 803–808.
- Nagy, V.V.; Cole, T.T.; Van Campenhout, C.; Khoung, T.T.; Leung, C.C.; Vermeiren, S.S.; Novatchkova, M.M.; Wenzel, D.D.; Cikes, D.D.; Polyansky, A.A.; et al. The evolutionarily conserved transcription factor PRDM12 controls sensory neuron development and pain perception. Cell Cycle 2015, 14, 1799–1808.
- Denk, F.; Bennett, D.; McMahon, S.B. Nerve Growth Factor and Pain Mechanisms. Annu. Rev. Neurosci. 2017, 40, 307–325.
- Bataille, A.; Leschiera, R.; L’Hérondelle, K.; Pennec, J.-P.; Le Goux, N.; Mignen, O.; Sakka, M.; Plée-Gautier, E.; Brun, C.; Oddos, T.; et al. In Vitro Differentiation of Human Skin-Derived Cells into Functional Sensory Neurons-Like. Cells 2020, 9, 1000.
- Kolomietz, E.; Marrano, P.; Yee, K.; Thai, B.; Braude, I.; Chun, K.; Minkin, S.; Kamel-Reid, S.; Minden, M.; Squire, J. Quantitative PCR identifies a minimal deleted region of 120 kb extending from the Philadelphia chromosome ABL translocation breakpoint in chronic myeloid leukemia with poor outcome. Leukemia 2003, 17, 1313–1323.
- Reid, A.G.; Nacheva, E.P. A potential role for PRDM12 in the pathogenesis of chronic myeloid leukaemia with derivative chromosome 9 deletion. Leukemia 2003, 18, 178–180.
- Huet, S.; Dulucq, S.; Chauveau, A.; Ménard, A.; Chomel, J.-C.; Maisonneuve, H.; Legros, L.; Perrin, M.-C.; Ferrant, E.; Moreilhon, C.; et al. Molecular characterization and follow-up of five CML patients with new BCR-ABL1 fusion transcripts. Genes Chromosom. Cancer 2015, 54, 595–605.
- Zhang, Y.; Yan, L.; Yao, W.; Chen, K.; Xu, H.; Ye, Z. Integrated Analysis of Genetic Abnormalities of the Histone Lysine Methyltransferases in Prostate Cancer. Med. Sci. Monit. 2019, 25, 193–239.
- Chen, X.; Wyler, S.C.; Li, L.; Arnold, A.G.; Wan, R.; Jia, L.; Landy, M.A.; Lai, H.C.; Xu, P.; Liu, C. Comparative Transcriptomic Analyses of Developing Melanocortin Neurons Reveal New Regulators for the Anorexigenic Neuron Identity. J. Neurosci. 2020, 40, 3165–3177.
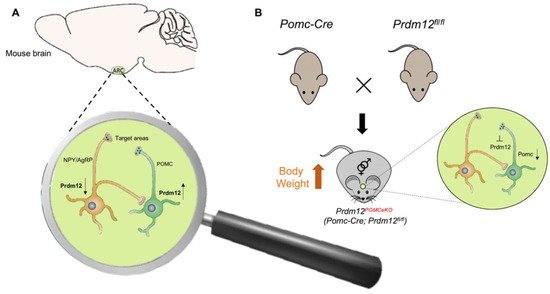
- Hael, C.E.; Rojo, D.; Orquera, D.P.; Low, M.J.; Rubinstein, M. The transcriptional regulator PRDM12 is critical for Pomc expression in the mouse hypothalamus and controlling food intake, adiposity, and body weight. Mol. Metab. 2020, 34, 43–53.

