Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thomas P. West | + 1937 word(s) | 1937 | 2021-11-01 03:33:30 | | | |
| 2 | Bruce Ren | Meta information modification | 1937 | 2021-11-09 02:44:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
West, T. Xylitol Biosynthesis in the Yeast Candida. Encyclopedia. Available online: https://encyclopedia.pub/entry/15805 (accessed on 24 July 2026).
West T. Xylitol Biosynthesis in the Yeast Candida. Encyclopedia. Available at: https://encyclopedia.pub/entry/15805. Accessed July 24, 2026.
West, Thomas. "Xylitol Biosynthesis in the Yeast Candida" Encyclopedia, https://encyclopedia.pub/entry/15805 (accessed July 24, 2026).
West, T. (2021, November 08). Xylitol Biosynthesis in the Yeast Candida. In Encyclopedia. https://encyclopedia.pub/entry/15805
West, Thomas. "Xylitol Biosynthesis in the Yeast Candida." Encyclopedia. Web. 08 November, 2021.
Copy Citation
Xylitol is an industrially important chemical due to its commercial applications. The use of xylitol as a sweetener as well as its utilization in biomedical applications has made it a high value specialty chemical.
xylitol
agricultural residues
grasses
xylose reductase
1. Introduction
The industrially important specialty chemical xylitol, whose annual commercial production is now approaching 40,000 tons [1], has several commercial applications (Figure 1). The highest value commercial application for xylitol use is as an alternate sweetener in such products as chewing gum and various foods including ice cream and candy [2]. Beyond its utilization as a sweetener, biomedical applications exist for xylitol, including being used to prevent ear inflammations and its ability to stimulate murine hybridoma cell production [3][4][5]. Xylitol is also used as a sugar substitute for diabetics [5]. With respect to human health, it has been reported that xylitol promotes the growth of beneficial bacteria by promoting the synthesis of propionate and short-chain fatty acids in the human colon [6]. The chemical synthesis of xylitol from xylose still remains the dominant production method of xylitol production [5][7]. The industrial process to synthesize xylitol involves the chemical hydrolysis of xylan followed by the hydrogenation of the resultant hemicellulose hydrolysate by catalysts including palladium and nickel [5][7]. During the chemical synthesis of xylitol, both high temperatures and high pressure are usually required, with typical temperatures being 80–140 °C and 8–10 MPa hydrogen pressure being employed [5]. These processes are highly energy intensive, which makes the production costs of synthesizing xylitol very expensive considering the high temperature, pressure and metal catalyst used for a sustained period of time [5]. Another problem with the chemical production of xylitol is the synthesis of side products such as arabitol by the chemical catalyst, which necessitates the purification of the xylitol being produced [5]. Clearly, the cost of producing xylitol by a large-scale process is going to be high, which makes this possible production process not feasible economically in the long term [7][8][9]. A more realistic approach for large-scale xylitol production would be to use the pentose sugars present in hydrolysates of low-value plant biomass [10]. Low-value biomass residues are excellent candidates for the large-scale production of xylitol. Such low-cost residues are readily available as a raw material for xylitol synthesis compared to the process components used during the chemical bioconversion of xylose into xylitol. This could include utilizing hydrolysates of agricultural residues or hydrolysates derived from various species of grasses [11][12][13][14][15][16]. It has been well documented that a number of hydrolysates from agricultural residues contain a high level of pentoses that could be used for bioconversion into xylitol. Similarly, it has been shown that a variety of hydrolyzed grasses support biobased xylitol production. One advantage of grasses is that they generally require low fertilizer input while producing high yields. With the fiber content of the grasses being relatively high, it has been demonstrated that both physical and enzymatic treatments of the grasses can result in a hydrolysate containing a high xylose concentration.
Figure 1. Structure of xylitol and its applications.
With hydrolysates containing high xylose levels, the bioconversion of the xylose present to xylitol can be accomplished by species of the yeast Candida. Prior work has identified a number of Candida species that are able to synthesize xylitol from the xylose present in hydrolyzed agricultural residues or grasses [17][18][19][20][21]. These yeast species contain the enzyme xylose reductase that primarily catalyzes the NADPH-dependent reduction of xylose to xylitol [1]. The resultant xylitol produced can be purified using activated carbon adsorption columns from the hydrolysate-containing medium [22].
2. Pathway of Xylitol Biosynthesis in the Yeast Candida
The ability of species of the yeast Candida to produce xylitol from xylose is due to the presence of the enzyme xylose reductase within their cells. Xylose reductase (EC 1.1.1.21) catalyzes the reduction of xylose by NADPH to the polyalcohol xylitol and NADP+ (Figure 2). It should be mentioned that some isolated yeast xylitol reductases can also use NADH as a cofactor. There is much interest in better understanding the xylose reductase structure to improve its efficiency in producing xylitol. Better understanding of yeast xylitol reductases could lead to the genetic engineering of this enzyme so that it has a greater affinity for its substrates, resulting in increased xylitol production. An engineered high activity xylitol reductase could efficiently synthesize xylitol from the high xylose-containing agricultural residues and grasses on an industrial scale. Xylose reductase has been investigated in a number of Candida species including Candida tenuis, Candida guillermondii, Candida parapsilosis, Candida intermedia and Candida tropicalis [23][24][25][26][27][28][29][30][31][32].
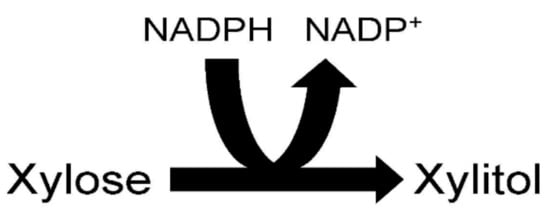
Figure 2. Xylose reductase reaction.
The xylose reductase from C. tenuis has been purified by dye ligand affinity chromatography and ion exchange chromatography to homogeneity [23]. Structurally, the purified enzyme was found to be a monomer or homodimer with each subunit having a molecular weight of 36,000–43,000 daltons [23][26]. This reductase from C. tenuis appears to differ from other family members of the aldo-keto reductase group, which exist purely as monomers. The isoelectric point of the reductase was determined to be 4.7 [23]. The reductase prefers NADPH compared to NADH as a cofactor based on binding specificity and it has been shown that NADP+ is a strong competitive inhibitor of the reaction [23]. The reductase was shown to contain a catalytic tetrad consisting of tyrosine, lysine, aspartic acid and histidine residues essential to its activity [26]. The lysine residue has been shown to be located near the coenzyme-binding site. Further, the presence of the epsilon amino group of the lysine residue in the reductase is thought to be a key element in its mode of catalysis [26]. In a pH 7.0-buffered assay incubated at 25 °C, the C. tenuis reductase had a Km and kcat of 87 mM and 18.2/s, respectively, for xylose. The reductase had a Km of 4.8 µM and 21.9/s, respectively, for NADPH, while it had a Km of 25.4 µM and kcat of 18.1/s, respectively, for NADH [23].
The xylose reductase gene fragment isolated from C. guilliermondii ATCC 20118 was cloned into Pichia pastoris GS115 [24]. This resulted in two xylose reductase activities being synthesized [24]. The xylose reductase activity, secreted extracellularly, was found to utilize only NADPH as its cofactor [24]. The molecular weight of this xylose reductase was shown to be 36,000 daltons [24]. The reductase was composed of 317 amino acids with the pI of the reductase shown to be 5.7 [24]. It was noted that the reductase was highly hydrophobic relative to amino acid composition including many leucine residues. It was also shown that three cysteine residues and seven histidine residues were present within the reductase structure similar to the P. pastoris xylose reductase [24]. In C. guilliermondii FTI 20037, a prior report characterized xylitol reductase in a crude extract prepared from cells cultured in a medium containing sugarcane bagasse hydrolysate [25]. The kinetics of the crude reductase were analyzed and it was observed that the xylose reductase had a Km of 64 mM for xylose and a Km of 9.5 µM for NADPH [25]. The optimal pH for the reductase was 5.5 and the enzyme was most stable when buffered at a pH between 5.0 and 5.5 [25]. The optimal temperature for the reductase was 65 °C and its enzyme activity was still stable after being heated to 60 °C for 10 min [25].
In C. intermedia, it was noted that the yeast contained two different forms of xylose reductase [27]. The isoforms of the enzyme were purified to homogeneity by a combination of affinity chromatography and ion exchange chromatography with relatively high yields. One form of the reductase was strictly specific for NADPH as a cofactor with a Km of 61 µM and a kcat of 14.6/s, while its Km and kcat for xylose were 82 mM and 14.6/s, respectively. The second form of the reductase was able to utilize either NADPH or NADH as a cofactor [27]. The Km of this reductase isoform for NADPH was 56 µM and its kcat was 11/s, while its Km for NADH was 28 µM and its kcat was 11.2/s. The Km of this reductase for xylose was about 50 mM and its kcat was about 10/s [27]. Structurally, both isoforms exist as homodimers with individual subunits having a molecular weight of 36,000 daltons. The isoelectric point of each isoform was found to differ with the NADPH-specific isoform having a pI of 4.38, while the second form had a pI of 4.59 [27]. It is interesting to note that the number of titratable cysteines in each isoform also differed. The NADPH-specific isoform had five titratable cysteines, while the second form had only two [27]. The inactivation of the cysteines in the NADPH-specific isoform resulted in a total loss of its activity [27]. It was thought that the ratio of available NADPH to NADH was a determining factor as to which isoform of the reductase was most active in the C. intermedia cells [27].
The xylitol reductase gene in C. parapsilosis was cloned in Escherichia coli and the reductase was purified to homogeneity using ion exchange chromatography, affinity chromatography and preparative electrophoresis [28]. The molecular weight of the C. parapsilosis reductase was 36,629 daltons and was composed of 324 amino acids. The reductase was shown to have a high catalytic efficiency for xylose as a substrate. The Km for xylose was calculated to be 31.5 mM, with its kcat being 46/s. Interestingly, this reductase preferred NADH as its cofactor instead of NADPH [28]. The Km of the reductase for NADH was 3.1 µM compared to the Km for NADPH being 36.5 µM [28]. The kcat of the reductase for NADH was 45.9/s relative to the kcat for NADPH being 4.6/s [28]. The optimal pH for the reductase was 6.0, while its temperature stability was greatest if stored at 4 °C. The reductase was subject to non-competitive inhibition by its product xylitol. The reaction mechanism for the C. parapsilosis reductase was thought to be an ordered sequential bi bi mechanism as has been proposed for other xylitol reductases [28].
In C. tropicalis, the properties of xylose reductase have been investigated [30][31][32]. The xylose reductase from C. tropicalis has been purified by cloning the xylitol reductase gene into Escherichia coli [30]. The purified reductase was crystalized and analyzed using X-ray diffraction [30]. The properties of a crude xylitol reductase activity isolated from a strain of C. tropicalis adapted to a hydrolysate of tree sawdust were studied [31]. The reductase was shown to be specific for NADP+ as its cofactor [31]. The reductase was still 95% active following 120 days at −80 °C. Further, the reductase was observed to be stable at a pH range between pH 5 and 7. The crude enzyme was stable for 24 h when incubated between 25 and 40 °C. The Km of the reductase was calculated to be 81.78 mM for xylose and 7.29 µM for NADPH [31]. The Vmax of the enzyme for xylose and NADPH was 178.57 µM/min and 12.5 µM/min, respectively [31]. The Km and Vmax values for the reductase were thought to be associated with the high rate of xylitol production noted by the strain. The molecular weight of the reductase was 36,600 daltons [30]. Another study using recombinant versions of the C. tropicalis reductase found that a serine residue at position 279 of its structure allowed increased catalysis compared to the presence of a leucine or an asparagine residue at position 279 [32]. The importance of a serine residue at position 279 of the reductase was thought to involve the binding of NADPH [32].
In summary, when comparing the previously investigated xylose reductases isolated from various species of Candida, there appear to be some similarities. First, the xylose reductases isolated from species of Candida appear to exhibit similar molecular weights. In general, the pH optimum of many of the characterized reductases from Candida species was similar. In addition, some of the reductases synthesized by the Candida species had a strict cofactor requirement for NADPH, while some synthesized a reductase activity that preferred NADPH as a cofactor but could still use NADH as a cofactor to catalyze the reaction. With respect to the kinetic properties of the Candida xylose reductases, the affinity of the enzymes for the nicotinamide cofactor was usually much greater than their affinity for xylose as a substrate.
References
- Gränstrom, T.B.; Izumori, K.; Leisola, M. A rare sugar xylitol. Part II: Biotechnological production and future applications of xylitol. Appl. Microbiol. Biotechnol. 2007, 74, 273–276.
- Edomi, A. Xylitol: Its properties and food applications. Food Technol. 1978, 32, 20–32.
- Petch, D.; Butler, M. The effect of alternative carbohydrates on the growth and antibody production of a murine hybridoma. Appl. Biochem. Biotechnol. 1996, 59, 93–104.
- Uhari, M.; Kontiokari, T.; Niemela, M. A novel use of xylitol sugar in preventing acute otitis media. Pediatrics 1998, 102, 879–884.
- Subroto, E.; Hayati, F. Chemical and biotechnological methods for the production of xylitol: A review. IJETER 2020, 8, 2508–2512.
- Xiang, S.; Ye, K.; Li, M.; Ying, J.; Wang, H.; Han, J.; Shi, L.; Xiao, J.; Shen, Y.; Feng, X.; et al. Xylitol enhances synthesis of propionate in the colon via cross-feeding of gut microbiota. Microbiome 2021, 9, 62.
- Xu, Y.; Chi, P.; Bilal, M.; Cheng, H. Biosynthetic strategies to produce xylitol: An economical venture. Appl. Microbiol. Biotechnol. 2019, 103, 5143–5160.
- Silva, S.S.; Matos, Z.R.; Carvalho, W. Effects of sulfuric acid loading and resident time on the composition of sugarcane bagasse hydrolysate and its use as a source of xylose for xylitol bioproduction. Biotechnol. Prog. 2005, 21, 1449–1452.
- Saha, B.C.; Kennedy, G.J. Production of xylitol from mixed sugars of xylose and arabinose without co-producing arabitol. Biocatal. Agric. Biotechnol. 2020, 29, 101786.
- Zhang, Y.-H.P. Reviving the carbohydrate economy via multiproduct lignocellulose biorefineries. J. Ind. Microbiol. Biotechnol. 2008, 35, 367–375.
- Mulkey, V.R.; Owens, V.N.; Lee, D.K. Management of warm-season grass mixtures for biomass production in South Dakota USA. Bioresour. Technol. 2008, 99, 609–617.
- West, T.P.; Peterson, J.L. Production of the polysaccharide curdlan by an Agrobacterium strain grown on a plant biomass hydrolysate. Can. J. Microbiol. 2014, 60, 53–56.
- West, T.P. Effect of nitrogen source concentration on curdlan production by Agrobacterium sp. ATCC 31749 grown on prairie cordgrass hydrolysates. Prep. Biochem. Biotechnol. 2016, 46, 85–90.
- West, T.P. Fungal production of the polysaccharide pullulan from a plant hydrolysate. Z. Naturforsch. C 2017, 72, 491–496.
- Kennedy, D.E., II; West, T.P. Effect of yeast extract addition to a mineral salts medium containing hydrolyzed plant xylan on fungal pullulan production. Z. Naturforsch. C 2018, 73, 319–323.
- West, T.P. Production of the polysaccharide curdlan by Agrobacterium species on processing coproducts and plant lignocellulosic hydrolysates. Fermentation 2020, 6, 16.
- Onishi, H.; Suzuki, T. Microbial production of xylitol from glucose. Appl. Microbiol. 1969, 18, 1031–1035.
- Barbosa, M.F.S.; de Medeiros, M.B.; de Mancilha, I.M.; Schneider, H.; Lee, H. Screening of yeasts for production of xylitol from D-xylose and some factors which affect xylitol yield in Candida guilliermondii. J. Ind. Microbiol. 1988, 3, 241–251.
- Mayerhoff, Z.D.V.L.; Roberto, I.C.; Silva, S.S. Xylitol production from rice straw hemicellulose using different yeast strains. Biotechnol. Lett. 1997, 19, 407–409.
- Guo, C.; Zhao, C.; He, P.; Shen, A.; Jiang, N. Screening and characterization of yeasts for xylitol production. J. Appl. Microbiol. 2006, 101, 1096–1104.
- Kwaka, S.; Job, J.H.; Yuna, E.J.; Jina, Y.-S.; Seo, J.-H. Production of biofuels and chemicals from xylose using native and engineered yeast strains. Biotechnol. Adv. 2019, 37, 219–283.
- Cardoso, B.S.; Forte, M.B.S. Purification of biotechnological xylitol from Candida tropicalis fermentation using activated carbon in fixed-bed adsorption columns with continuous feed. Food Bioprod. Process. 2021, 126, 73–80.
- Neuhauser, W.; Haltrich, D.; Kulbe, K.D.; Nideetzky, B. NAD(P)H-dependent aldose reductase from the xylose-assimilating yeast Candida tenuis: Isolation, characterization and biochemical properties of the enzyme. Biochem. J. 1997, 326, 683–692.
- Handumrongkul, C.; Ma, D.-P.; Silva, J.L. Cloning and expression of Candida guilliermondii xylose reductase gene (xyl1) in Pichia pastoris. Appl. Microbiol. Biotechnol. 1998, 49, 399–404.
- Sene, L.; Felipe, M.G.A.; Silva, S.S.; Vitolo, M. Preliminary kinetic characterization of xylose reductase and xylitol dehydrogenase extracted from Candida guilliermondii FTI 20037 cultivated in sugarcane bagasse hydrolysate for xylitol production. Appl. Biochem. Biotechnol. 2001, 91–93, 671–680.
- Kavanagh, K.L.; Klimacek, M.; Nidetzky, B.; Wilson, D.K. The Structure of apo and holo forms of xylose reductase, a dimeric aldo-keto reductase from Candida tenuis. Biochemistry 2002, 41, 8785–9795.
- Mayr, P.; Bruggler, K.; Kulbe, K.D.; Nidetzky, B. D-Xylose metabolism by Candida intermedia: Isolation and characterisation of two forms of aldose reductase with different coenzyme specificities. J. Chromatogr. B Biomed. Sci. Appl. 2000, 737, 195–202.
- Lee, J.-K.; Koo, B.S.; Kim, S.-Y. Cloning and characterization of the xyl1 gene, encoding an NADH-preferring xylose reductase from Candida parapsilosis, and its functional expression in Candida tropicalis. Appl. Environ. Microbiol. 2003, 69, 6179–6188.
- Kratzer, R.; Wilson, D.K.; Nidetzky, B. Catalytic mechanism and substrate selectivity of aldo-keto reductases: Insights from structure-function studies of Candida tenuis xylose reductase. Life 2006, 58, 499–507.
- Chen, L.-C.; Huang, S.-C.; Chuankhayan, P.; Chen, C.-D.; Huang, Y.-H.; Jeyakanthan, J.; Pang, H.-F.; Men, L.-C.; Chen, Y.-C.; Wang, Y.-K.; et al. Purification, crystallization and preliminary X-ray crystallographic analysis of xylose reductase from Candida tropicalis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, F65, 419–421.
- Rafiqul, I.S.M.; Sakinah, A.M.M. Biochemical properties of xylose reductase prepared from adapted strain of Candida tropicalis. Appl. Biochem. Biotechnol. 2015, 175, 387–399.
- Kim, S.; Lee, J.; Sung, B.H. Isolation and characterization of the stress-tolerant Candida tropicalis YHJ1 and evaluation of its xylose reductase for xylitol production from acid pre-treatment wastewater. Front Bioeng. Biotechnol. 2019, 7, 138.
More
Information
Subjects:
Biotechnology & Applied Microbiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
09 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No