 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Davide Bassetti | + 1128 word(s) | 1128 | 2021-07-14 09:00:17 | | | |
| 2 | Amina Yu | -29 word(s) | 1099 | 2021-11-09 04:04:35 | | |
Video Upload Options
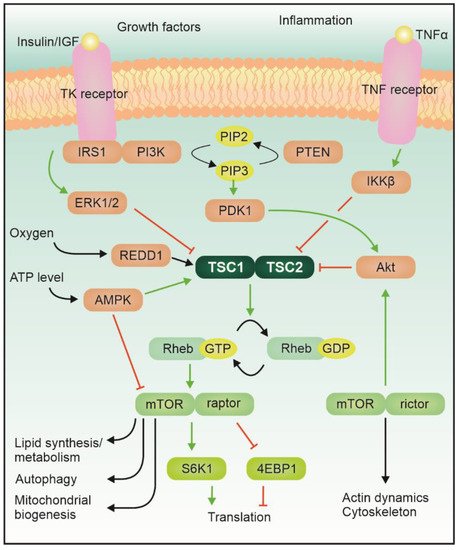
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disease featuring localized uncontrolled growth of tissues, known as hamartomas, in many organs, including the central nervous system (CNS). A large proportion of the cases are sporadic mutations (ca. two thirds). The disease results from a loss-of-function mutation in either the TSC1 or TSC2 gene, which encode for hamartin and tuberin proteins, respectively; two proteins that together form the Tsc1–Tsc2 complex. This complex, which is the converging center of several signaling pathways, has GTPase activity and can inhibit the activity of Ras homolog enriched in the brain (Rheb). The complex itself is modulated by phosphorylation via, for example, Akt or AMPK (Figure 1).
1. Introduction
The mTOR protein itself is a serine/threonine kinase which acts as an integrator of various signals belonging to different modalities, related for example to the energetic status of the cell and the availability of growth factors and nutrients, and regulates the growth program of the cell [1]. Activation of mTORC1 leads to phosphorylation of its main downstream targets (including 4e binding protein 1 and S6 kinase), which control local protein synthesis, cellular growth, and autophagy [2]. mTORC2, instead, mostly regulates actin cytoskeleton dynamics via PKC or small GTPases of the Rho family. Albeit the role of mTORC2 in other diseases featuring mTOR hyperactivation is becoming more prominent, in TSC its contribution remains elusive [3].
In the central nervous system, the mTOR pathway is crucial for a variety of functions, including cell growth, local protein synthesis, synaptogenesis and synaptic pruning. Alteration of mTOR activity results in neurological effects, like epilepsy, intellectual disability, and autism spectrum disorder (ASD) (for review, see [4][5][6]). Additionally, TSC patients often report a number of other conditions including overactivity, impulsivity, anxiety, mood swings, aggressivity, and less often self-injury, obsessions and psychosis. This set of further symptoms takes the collective name of Tuberous-sclerosis Associated Neuropsychiatric Disorders (TANDs, see [7]).
Homozygous mutations in either Tsc1 or Tsc2 are not viable [8][9]. Heterozygous Tsc1 and Tsc2 mice do not display brain lesions, and Tsc2+/− rats feature cortical tubers, albeit with low incidence [10][11]. No spontaneous seizures were detected in these models, beside in Tsc1+/−, which undergoes a transient period of epileptiform activity at young age [12]. While all three models present with varying degrees of behavioral and cognitive deficits [13][14][15][16][17][18][19][20], their relationship with the rich variety of manifestations observed in ASD and TANDs symptoms in humans is not necessarily straightforward.
Pharmacological induction of seizures during early developmental stages (P7 and P14) in WT rats causes the appearance of social interaction deficits at 3 to 6 months of age. These results suggest that both haploinsufficiency of Tsc2 without epileptic activity and transient epileptic seizures during early postnatal development can lead to ASD symptoms in later life [19].
2. Synaptic transmission
As Tsc1–Tsc2-mediated changes in mTOR activity affect both axon formation and guidance, i.e., the presynaptic site, as well as spine shape and stabilization, i.e., the postsynaptic site, one can expect that Tsc1–Tsc2 deletion can influence synaptic transmission.
Pyramidal neurons in the medial prefrontal cortex (mPFC) of Tsc2+/−mice demonstrated no difference in the amplitude of AMPA-mediated mEPSCs as compared with controls, while mEPSC frequency was increased at P15–P40, suggesting a potentiation of glutamatergic drive in this mouse model [21][22]. In contrast to the mPFC, a decrease in both mEPSC amplitude and mEPSC frequency has been reported in the somatosensory cortex of Tsc2+/−mice around P15. At the same time, eEPSC amplitudes were not significantly different, indicating constant strength of the glutamatergic drive [23].
Activation of both NMDA and mGluR receptors can contribute to induction of long-term potentiation (LTP) and/or long-term depression (LPD), and indeed changes in synaptic plasticity were reported in Tsc1–Tsc2 deficient mice and rats. Levels of both LTP and LTD induced in acute slices by different stimulation patterns were significantly reduced in Tsc2-deficient rats [24]. Interestingly, using the same mouse model, Ehninger and colleagues reported no change in paired-pulse plasticity and early-LTP strength, but the level of late-LTP induced via tetanic stimulation was higher in Tsc2+/−mice as compared to controls [15]. As for the changes in glutamatergic synaptic transmission, also plasticity shows a complex picture of developmental and region-specific regulation.
An increase in excitation could have similar functional consequences as a decrease in inhibition, i.e., leading to increased noise in key circuits responsible for computation [25]. In mice with selective genetic inactivation of Tsc1 in a subset of hippocampal CA1 neurons in vivo featured strongly elevated mEPSC frequency [26]. The Tsc-deficient cells however displayed reduced intrinsic excitability which could effectively shunt the effects of increased excitatory synaptic transmission on the network activity. Based on this, the authors proposed that the increased glutamatergic synaptic transmission could have actually been a secondary effect, and the observed increase in the E/I ratio reflected rather a reduction in inhibitory transmission, showing how increased activity in mTOR pathway can lead to a cascade of changes and consequent adjustments [27].
The existence of multiple critical time-windows has been directly shown while investigating the effects of rapamycin on dendrite arborization and spine number. By the end of the first postnatal month Tsc1-suppressed cells showed more complex dendrite branching and higher spine density. Interestingly, dendrite arborization was normalized by rapamycin treatment between P1 and P13 but not after P15, while abnormal spine stabilization was achieved by rapamycin treatment between P15 and P28, but not before P13 [28]. Similarly, Tsc1+/−mice showed spontaneous epileptic seizures only transiently in a specific time window (before P19).
Using electrophysiological recordings of postsynaptic currents functional I ratio was elevated in the mPFC, but arising from selective potentiation of glutamatergic transmission [21]. In contrast to the hyperexcitability observed before P19 in Tsc1+/−mice [12], sensory-evoked spiking was not increased in Tsc2+/−mice. The elevated E/I ratio might represent a compensatory tuning to stabilize the neuronal network [23].
As extracellular GABA concentration is regulated locally by GABA transporters, astrocytes could play a pivotal role in the tonic modulation of glutamatergic and GABAergic synaptic transmission. In fact, WT neurons cultured in astrocyte-conditioned medium of astrocytes derived from TSC-patient stem cells showed an increased number of GABAergic synapses [29]. For instance, deletion of either Tsc1 or Tsc2 in differentiated astrocytes did not produce tuber-like lesions (and nevertheless resulted in seizures), but inactivation of Tsc1 or Tsc2 in undifferentiated radial glia cells early during development resulted in phenotypic neuropathological features characteristic for TSC patients, including aberrations in cortical cytoarchitecture as well as epileptic seizures ([30], for review [31]).
References
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976.
- Ben-Sahra, I.; Manning, B.D. MTORC1 Signaling and the Metabolic Control of Cell Growth. Curr. Opin. Cell Biol. 2017, 45, 72–82.
- Chen, C.-J.; Sgritta, M.; Mays, J.; Zhou, H.; Lucero, R.; Park, J.; Wang, I.-C.; Park, J.H.; Kaipparettu, B.A.; Stoica, L.; et al. Therapeutic Inhibition of MTORC2 Rescues the Behavioral and Neurophysiological Abnormalities Associated with Pten-Deficiency. Nat. Med. 2019, 25, 1684–1690.
- Bockaert, J.; Marin, P. MTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187.
- Sato, A. MTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol. Disord.-Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2016, 15, 533–543.
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular Neurobiology of MTOR. Neuroscience 2017, 341, 112–153.
- de Vries, P.J.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’Amato, L.; d’Augères, G.B.; Ferreira, J.C.; et al. TSC-Associated Neuropsychiatric Disorders (TAND): Findings from the TOSCA Natural History Study. Orphanet J. rare Dis. 2018, 13, 157.
- Rennebeck, G.; Kleymenova, E.V.; Anderson, R.; Yeung, R.S.; Artzt, K.; Walker, C.L. Loss of Function of the Tuberous Sclerosis 2 Tumor Suppressor Gene Results in Embryonic Lethality Characterized by Disrupted Neuroepithelial Growth and Development. Proc. Natl. Acad. Sci. USA 1998, 95, 15629–15634.
- Kobayashi, T.; Minowa, O.; Sugitani, Y.; Takai, S.; Mitani, H.; Kobayashi, E.; Noda, T.; Hino, O. A Germ-Line Tsc1 Mutation Causes Tumor Development and Embryonic Lethality That Are Similar, but Not Identical to, Those Caused by Tsc2 Mutation in Mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8762–8767.
- Mizuguchi, M.; Takashima, S.; Yamanouchi, H.; Nakazato, Y.; Mitani, H.; Hino, O. Novel Cerebral Lesions in the Eker Rat Model of Tuberous Sclerosis: Cortical Tuber and Anaplastic Ganglioglioma. J. Neuropathol. Exp. Neurol. 2000, 59, 188–196.
- Takahashi, D.K.; Dinday, M.T.; Barbaro, N.M.; Baraban, S.C. Abnormal Cortical Cells and Astrocytomas in the Eker Rat Model of Tuberous Sclerosis Complex. Epilepsia 2004, 45, 1525–1530.
- Lozovaya, N.; Gataullina, S.; Tsintsadze, T.; Tsintsadze, V.; Pallesi-Pocachard, E.; Minlebaev, M.; Goriounova, N.A.; Buhler, E.; Watrin, F.; Shityakov, S.; et al. Selective Suppression of Excessive GluN2C Expression Rescues Early Epilepsy in a Tuberous Sclerosis Murine Model. Nat. Commun. 2014, 5.
- Sato, A.; Kasai, S.; Kobayashi, T.; Takamatsu, Y.; Hino, O.; Ikeda, K.; Mizuguchi, M. Rapamycin Reverses Impaired Social Interaction in Mouse Models of Tuberous Sclerosis Complex. Nat. Commun. 2012, 3, 1292.
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of MTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143.
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D.J.; Ramesh, V.; Silva, A.J. Reversal of Learning Deficits in a Tsc2+/− Mouse Model of Tuberous Sclerosis. Nat. Med. 2008, 14, 843–848.
- Ehninger, D.; Silva, A.J. Increased Levels of Anxiety-Related Behaviors in a Tsc2 Dominant Negative Transgenic Mouse Model of Tuberous Sclerosis. Behav. Genet. 2011, 41, 357–363.
- Goorden, S.M.I.; van Woerden, G.M.; van der Weerd, L.; Cheadle, J.P.; Elgersma, Y. Cognitive Deficits in Tsc1+/−mice in the Absence of Cerebral Lesions and Seizures. Ann. Neurol. 2007, 62, 648–655.
- Haji, N.; Riebe, I.; Aguilar-Valles, A.; Artinian, J.; Laplante, I.; Lacaille, J.-C. Tsc1 Haploinsufficiency in Nkx2.1 Cells Upregulates Hippocampal Interneuron MTORC1 Activity, Impairs Pyramidal Cell Synaptic Inhibition, and Alters Contextual Fear Discrimination and Spatial Working Memory in Mice. Mol. Autism 2020, 11, 29.
- Waltereit, R.; Japs, B.; Schneider, M.; de Vries, P.J.; Bartsch, D. Epilepsy and Tsc2 Haploinsufficiency Lead to Autistic-Like Social Deficit Behaviors in Rats. Behav. Genet. 2011, 41, 364–372.
- Young, D.M.; Schenk, A.K.; Yang, S.-B.; Jan, Y.N.; Jan, L.Y. Altered Ultrasonic Vocalizations in a Tuberous Sclerosis Mouse Model of Autism. Proc. Natl. Acad. Sci. USA 2010, 107, 11074–11079.
- Bassetti, D.; Lombardi, A.; Kirischuk, S.; Luhmann, H.J. Haploinsufficiency of Tsc2 Leads to Hyperexcitability of Medial Prefrontal Cortex via Weakening of Tonic GABAB Receptor-Mediated Inhibition. Cereb. Cortex 2020.
- Bassetti, D.; Luhmann, H.J.; Kirischuk, S. Presynaptic GABA(B) Receptor-Mediated Network Excitation in the Medial Prefrontal Cortex of Tsc2+/− Mice. Pflugers Arch. 2021, in press.
- Antoine, M.W.; Langberg, T.; Schnepel, P.; Feldman, D.E. Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 2019, 101, 648–661.e4.
- von der Brelie, C.; Waltereit, R.; Zhang, L.; Beck, H.; Kirschstein, T. Impaired Synaptic Plasticity in a Rat Model of Tuberous Sclerosis. Eur. J. Neurosci. 2006, 23, 686–692.
- Rubenstein, J.L.R.; Merzenich, M.M. Model of Autism: Increased Ratio of Excitation/Inhibition in Key Neural Systems. Genes Brain Behav. 2003, 2, 255–267.
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of Tsc1 In Vivo Impairs Hippocampal MGluR-LTD and Increases Excitatory Synaptic Function. J. Neurosci. 2011, 31, 8862–8869.
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/Inhibitory Synaptic Imbalance Leads to Hippocampal Hyperexcitability in Mouse Models of Tuberous Sclerosis. Neuron 2013, 78, 510–522.
- Cox, R.L.; Calderon de Anda, F.; Mangoubi, T.; Yoshii, A. Multiple Critical Periods for Rapamycin Treatment to Correct Structural Defects in Tsc-1-Suppressed Brain. Front. Mol. Neurosci. 2018, 11.
- Dooves, S.; van Velthoven, A.J.H.; Suciati, L.G.; Heine, V.M. Neuron–Glia Interactions in Tuberous Sclerosis Complex Affect the Synaptic Balance in 2D and Organoid Cultures. Cells 2021, 10, 134.
- Magri, L.; Cominelli, M.; Cambiaghi, M.; Cursi, M.; Leocani, L.; Minicucci, F.; Poliani, P.L.; Galli, R. Timing of MTOR Activation Affects Tuberous Sclerosis Complex Neuropathology in Mouse Models. Dis. Models Mech. 2013, 6, 1185–1197.
- Zimmer, T.S.; Broekaart, D.W.M.; Gruber, V.-E.; van Vliet, E.A.; Mühlebner, A.; Aronica, E. Tuberous Sclerosis Complex as Disease Model for Investigating MTOR-Related Gliopathy During Epileptogenesis. Front. Neurol. 2020, 11, 1028.

