 +1 credit
+1 credit
Video Upload Options
Deubiquitinase (DUB) is an essential component in the ubiquitin-proteasome system (UPS) by removing ubiquitin chains from substrates, thus modulating the expression, activity, and localization of many proteins that contribute to tumor development and progression. DUBs have emerged as promising prognostic indicators and drug targets. DUBs have shown significant roles in regulating breast cancer growth, metastasis, resistance to current therapies, and several canonical oncogenic signaling pathways. In addition, specific DUB inhibitors have been identified and are expected to benefit breast cancer patients in the future.
1. Introduction
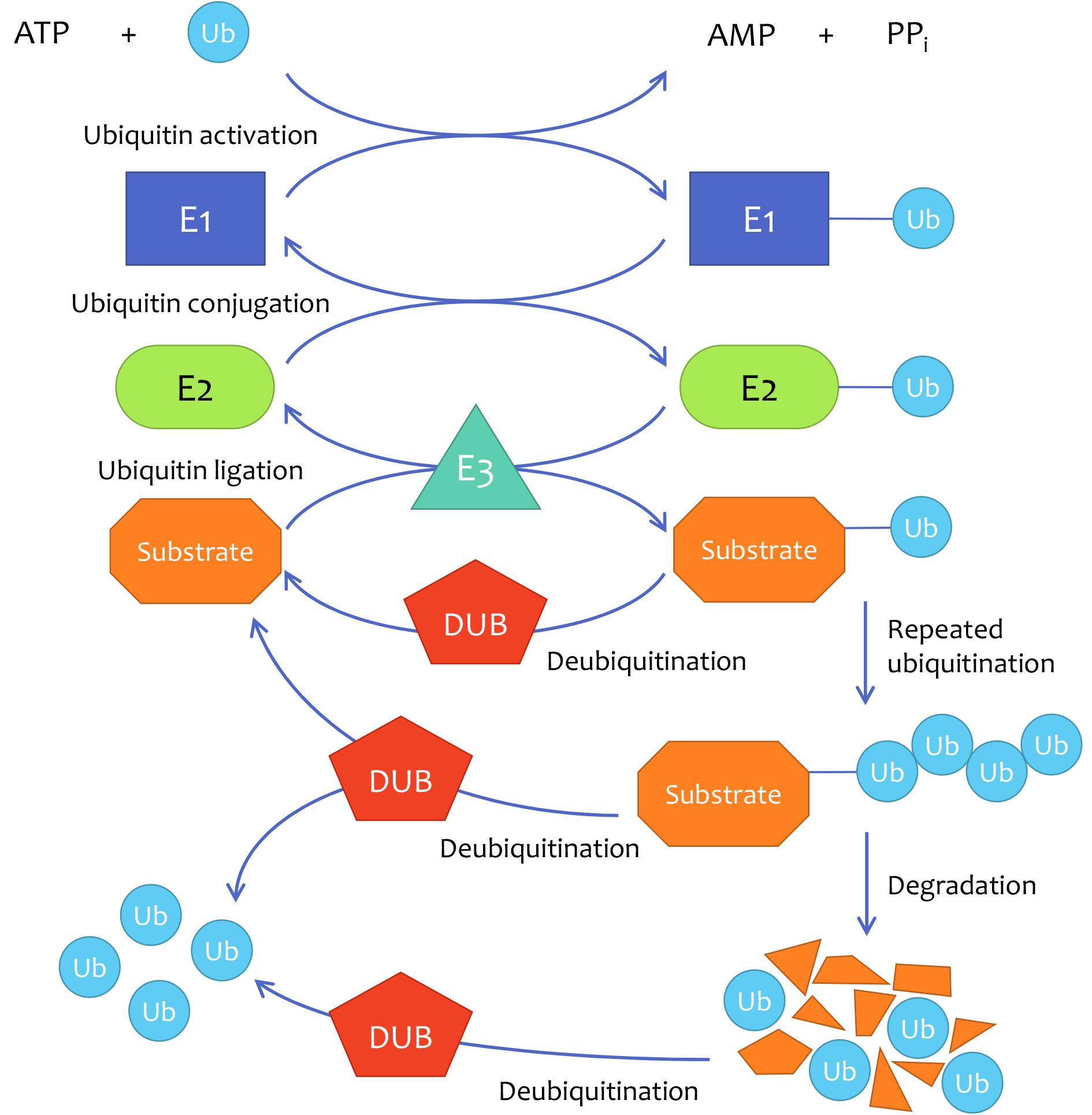
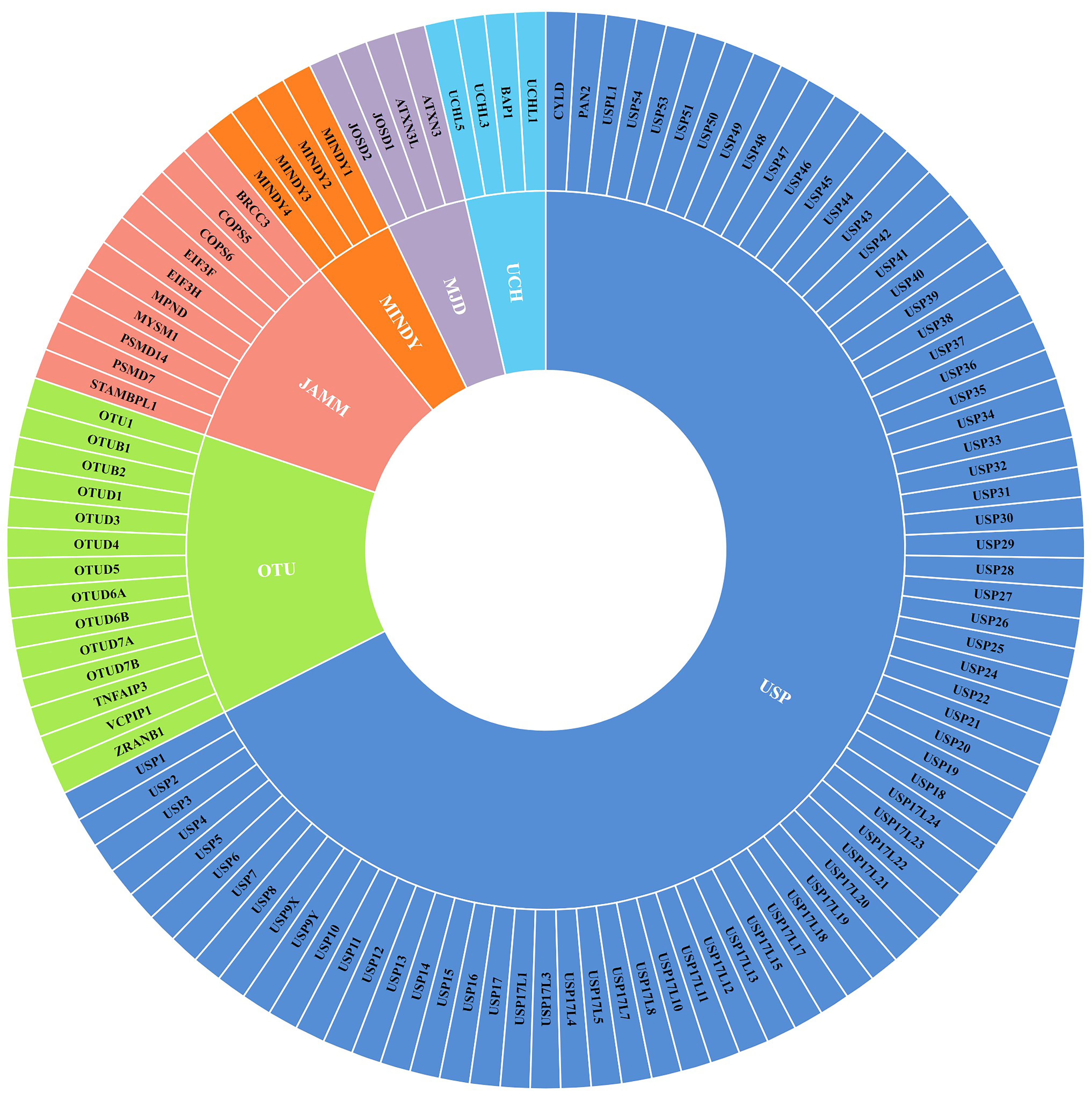
2. DUBs in Breast Cancer Growth
2.1. DUBs of c-Myc
2.2. DUBs of KLF5
2.3. DUBs That Regulate H2B Monoubiquitination Levels
2.4. DUBs of Cell Cycle Regulatory Components
2.5. Other DUBs in Breast Cancer Growth
3. DUBs in Breast Cancer Metastasis
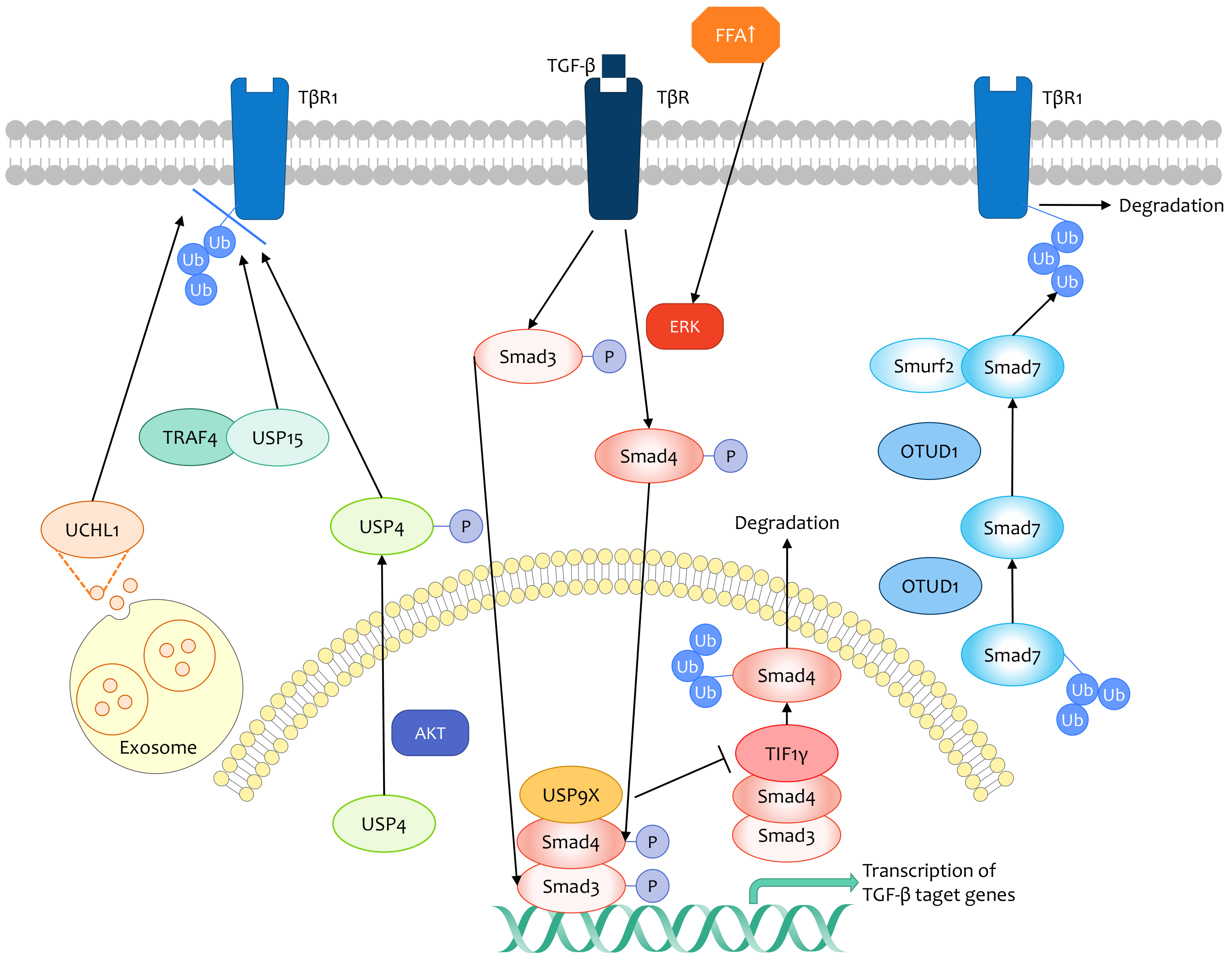
3.1. TGF-β Signaling Pathway
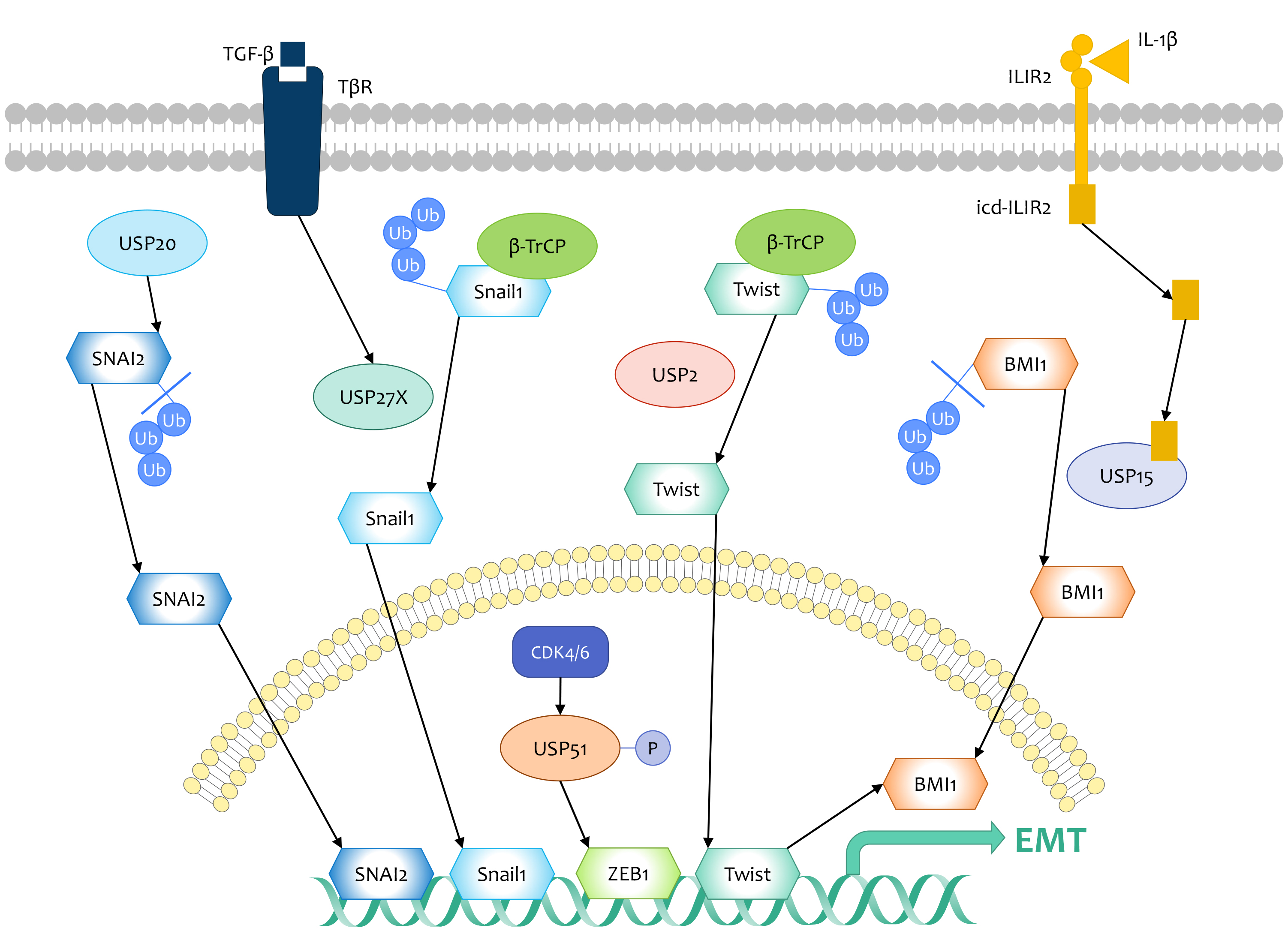
3.2. DUBs That Target EMT Regulators
3.3. Other DUBs Regulating Breast Cancer Metastasis
4. DUBs in Immunosuppression of Breast Cancer
Although cancer cells express antigens that can be recognized by T cells and activate the immune system[87], most tumors escape from immune surveillance through various mechanisms, including self-modification of cancer cells and alteration of tumor microenvironment. For instance, cancer cells highly express programmed death ligand 1 (PD-L1), bind with its receptor, and downregulate the activation of immune responses induced by T cells[88].
CSN5 was identified as a critical component in PD-L1-mediated immune evasion that inhibits PD-L1 poly-ubiquitination and protects it from proteasomal degradation. It is found that CSN5 is upregulated transcriptionally by NF-κB activation of p65[89]. Additionally, lncRNA also functions as an upstream signal to regulate CSN5. LncRNA GATA3-AS1 enhances CSN5 expression via separation of miR-676-3p from CSN5, thus contributing to the immune escape of TNBC cells[90]. According to clinical evidence, the level of CSN5 is positively related with PD-L1 in breast cancer tissues, and overexpression of CSN5 indicates poor prognosis in patients with breast cancers[89].
Recently, OTUB1 has been found as a novel DUB of PD-L1 in breast cancer. OTUB1 stabilizes PD-L1 and protects it from endoplasmic reticulum-associated degradation (ERAD) by removing its K48-linked ubiquitin chains. Consistently, loss of OTUB1 leads to PD-L1 reduction in breast cancer cells, enhancing their sensitivity to the cytotoxicity of immune cells[91].
In conclusion, the NF-κB/p65/CSN5/PD-L1, GATA3-AS1/miR-676-3p/CSN5/PD-L1, and OTUB1/PD-L1 axis promotes the immunosuppression of breast cancer.
5. DUBs in Chemoresistance and Chemosensitivity of Breast Cancer
The high incidence of breast cancer patients relapsing after chemotherapy indicates that breast cancer cells have complex mechanisms of chemoresistance.
5.1. Tamoxifen (SERM)
Generally, breast cancer is an estrogen-dependent malignancy. Consequently, chemotherapy with tamoxifen, a representative drug of estrogen antagonists, possesses a good therapeutic effect on patients with breast cancer, and changes in the ERα signaling pathway intensify the tendency of endocrine resistance[92].
USP22 deubiquitinates and stabilizes ERα, enhancing ERα-induced transactivation in breast cancer cells. At the molecular level, USP22 is demonstrated as a coactivator of downstream genes, which interacts with the cis-acting element together with ERα. As a result, USP22 increases breast cancer resistance to ERα antagonists. In breast cancer cell lines, USP22 reduction enhances the inhibitory effects on proliferation of ERα antagonist ICI 182,780 and tamoxifen by increasing cell sensitivity to endocrine therapy[22].
USP1 is also an essential deubiquitinase in ERα signaling, which enhances ERα stability through cleaving its Lys48-linked ubiquitin chains. According to TCGA and KMPLOT databases, high expression of USP1 is relevant to poor prognosis in ERα+ breast cancer patients[93].
Knockdown of USP9X gives rise to tamoxifen resistance by enhancing ERα’s interaction with chromatin. Although there is a physical interaction between USP9X and ERα, ERα is not the direct substrate for USP9X, indicating USP9X may deubiquitinate ERα cofactors to regulate ERα binding with chromatin[94].
It is found that the epidermal growth factor receptor (EGFR) represses ERα transcription via hyperactivation of MAPK signaling[95]. In addition, UCHL1 downregulates ERα by deubiquitinating and stabilizing EGFR, thus increasing tamoxifen resistance in ERα- breast cancer. UCHL1 inhibition offers a novel treatment for breast cancer patients with ERα shortage and decrease[96].
5.2. Enzalutamide (Antiandrogen)
According to the results of tissue microarrays from 3093 patients, 77% of invasive breast carcinomas are androgen receptor (AR) positive, indicating AR is frequently expressed in breast tumors[97]. The AR pathway is critical in AR+ breast cancer, functionally interacting with multiple classic oncogenic signaling pathways. Importantly, AR-targeted therapies, including the AR antagonist, enzalutamide, have been demonstrated to be effective against breast cancer[98]. USP14 is required for enhancing AR+ breast cancer cell proliferation through deubiquitination and stabilization of AR[99]. Moreover, USP14 expression has a positive correlation with AR expression according to the results from the TCGA database and is remarkably high in all subtypes of breast cancer. Thus, USP14 promotes resistance to enzalutamide in AR+ breast cancer[100].
5.3. Genotoxic Agents
Genotoxic agents such as doxorubicin (Dox)[101], irinotecan (CPT-11)[102], and cisplatin[103], are regarded as conventional treatments for breast cancer patients.
OTULIN, a member of OTU family, selectively recognizes and removes linear polyubiquitin chains from proteins[104]. OTULIN enhances TNBC resistance to Dox and CPT-11 through activation of the Wnt/β-catenin pathway, which contributes to chemoresistance by maintaining CSCs. Mechanistically, DNA damage promotes c-Abl translocation from nuclear to cytoplasm, where c-Abl promotes OTULIN phosphorylation at Tyr56. Then, OTULIN prompts the Wnt/β-catenin pathway by attenuating the linear ubiquitination of β-catenin and facilitates breast cancer cells alteration to a chemoresistant state. Moreover, clinical data show that increased levels of OTULIN and β-catenin significantly correlate with poor prognosis and chemoresistance in TNBC patients[105].
According to the results of the viability of different breast cancer cell lines after cisplatin treatment, ER- breast cancer is more resistant to cisplatin[106]. The deubiquitinase USP9X stabilizes MCL1, whose overexpression contributes to chemoresistance and poor prognosis in breast cancer[107]. Downregulation of USP9X reinforces cisplatin sensitivity in ER- breast cancer cells, which is speculated to be a result of the degradation of MCL1[106].
C-Jun activation domain-binding protein-1 (Jab1), also known as CSN5, which is negatively regulated and directly targeted by miR-17, increases cisplatin resistance in TNBC[108]. Jab1 also contributes to cellular resistance to cisplatin by enhancing Rad51 activity in DNA damage repair with the assistance of p53[109].
EMT transcription factors are significant for the acquisition of chemoresistance in cancer cells. For example, radiation or chemotherapy induces the expression of the Snail/Slug family in ovarian cancers. This, in turn, enhances cell survival by weakening the expression of the p53-mediated apoptotic gene and derepressing the expression of self-renewal genes[110]. Similarly, Snail1 may contribute to chemoresistance in breast cancer patients following the above-mentioned regulation. It has been demonstrated that USP27X is a putative deubiquitinase for Snail1, which enhances breast cancer cells resistance to cisplatin via stabilizing Snail1 and at least reinforcing repression of apoptosis-associated genes[75].
5.4. PARPi
BRCA1/2 are key components in the process of homologous recombination (HR) targeting the repair of DNA double-strand breaks (DSBs). Additionally, Poly-(ADP-ribose) polymerase (PARP) functions as a critical enzyme for DNA single-strand breaks repair, making PARP inhibitors (PARPi) an effective therapeutic strategy for cancer patients with BRCA mutations[111]. Therefore, it is necessary to find valid biomarkers identifying breast cancers that are sensitive to PARPi treatment.
A study found that USP15 affects breast cancer cell sensitivity to PARPi via regulation of HR. MDC1 recruits USP15 to DNA damage sites, where the BRCT domain of BARD1 is deubiquitinated by USP15, thereby enhancing BRCA1/BARD1 retention that facilitates DSB end resection. Investigators also speculated that breast cancer patients with USP15 M861V and D967H mutants are more sensitive to PARPi treatment, suggesting that these two sites contribute to the interaction with BARD1[112].
Moreover, BRCA2 recruits Rad51 to DSBs in the HR repair pathway to catalyze homologous pairing[113]. In addition, the deubiquitinase activity of UCHL3 is essential in this process. Mechanistically, ATM activates UCHL3 after DNA damage, which in turn enhances Rad51 interaction with BRCA2 via deubiquitination. Thus, UCHL3 strengthens the HR signaling pathway in DNA repair, rendering breast cancer cells resistant to PARPi. Likewise, according to clinical cases, UCHL3 overexpression functions as a prognostic index for unfavorable outcomes in breast cancer patients[114].
RNF169 is an atypical regulator in DSB repair that augments the accurate HR pathway instead of the nonhomologous end-joining (NEHJ) pathway[115]. USP7 interacts with RNF169 by UBL domains, then deubiquitinates and stabilizes RNF169, which effectively accumulates at DSBs in the promotion of HR. As a result, the USP7–RNF169 axis contributes to accurate DSB repair and facilitates breast cancer cells resistance to PARPi[116].
6. DUBs in Radioresistance and Radiosensitivity of Breast Cancer
Radiation therapy has increasingly become critical and conventional in breast cancer management. However, the presence of radioresistant cancer cells makes patients suffer from local tumor recurrences. It is therefore important to observe factors involve in radioresistance and explore potent tumor radiosensitizers.
It is well known that cancer stem cells (CSCs) are able to prompt cell cycle checkpoints, thus leading to radioresistance in tumors[117]. Meanwhile, EMT enables cells to obtain stem-like properties, indicating that EMT engages in radioresistance. Researchers identified that ZEB1, a core factor of EMT, is amplified in radioresistant subtypes of breast cancer. ZEB1 is phosphorylated by ATR, a component of the DNA damage repair (DDR) pathway. Then, ZEB1 combines with USP7 to increase its deubiquitinase and stabilization ability towards checkpoint kinase 1 (CHK1), thus facilitating the HR pathway that contributes to radioresistance[118]. In addition to EMT transcription factor ZEB1, long noncoding RNA LINC02582 also promotes radioresistance through interacting with USP7 and stabilizing CHK1. LINC02582 functionally serves as a molecular target of miR-200c, which has been previously demonstrated as a radiosensitizer in breast cancer[119]. PHF8 is also identified as another substrate of USP7, which involves in DSB repair via recruitment of BLM and KU70[31]. In conclusion, interfering USP7 deubiquitinase activity elevates breast cancer sensitivity to radiation therapy.
Rad51, a component in DNA repair pathway, is regarded as a selective target to sensitize tumors to cytotoxic treatments[120]. It is found that UCHL3 weakens radiosensitivity in breast cancer cells by deubiquitinating and activating Rad51. Interventions targeting UCHL3 may improve the curative effect in combination with radiation treatment[114].
USP52 stabilizes the histone chaperone ASF1A by removing K48-linked polyubiquitin chains, then ASF1A delivers classical S-phase histones H3.1-H4 dimer to replication-coupled chromatin. Therefore, USP52-mediated ASF1A deubiquitination is essential in sustaining genome stability upon DNA damage. Analysis of breast cancer cell viability showed that the USP52/ASF1A signaling promotes tumor cells resistance to ionizing radiation[121].
Moreover, the UCHL1/HIF-1 axis plays an important role in promoting breast cancer resistance to radiotherapy. UCHL1 upregulates the activity of HIF-1 via deubiquitination of its subunit HIF-1α. Then, HIF-1 activates reprogramming of glucose metabolism and the subsequent pentose phosphate pathway (PPP), thus increasing the level of reduced glutathione (GSH). It is widely recognized that intracellular antioxidants represented by GSH protect cancer cells from radiation-induced DNA lesions through scavenging free radicals and other oxidative products[122].
In summary, the ATM/ZEB1/USP7/CHK1, miR-200c/LINC02582/USP7/CHK1, USP7/PHF8, UCHL3/RAD51, USP52/ASF1A, and UCHL1/HIF-1 signaling axis are potential targets to improve the radiosensitivity of breast cancer.
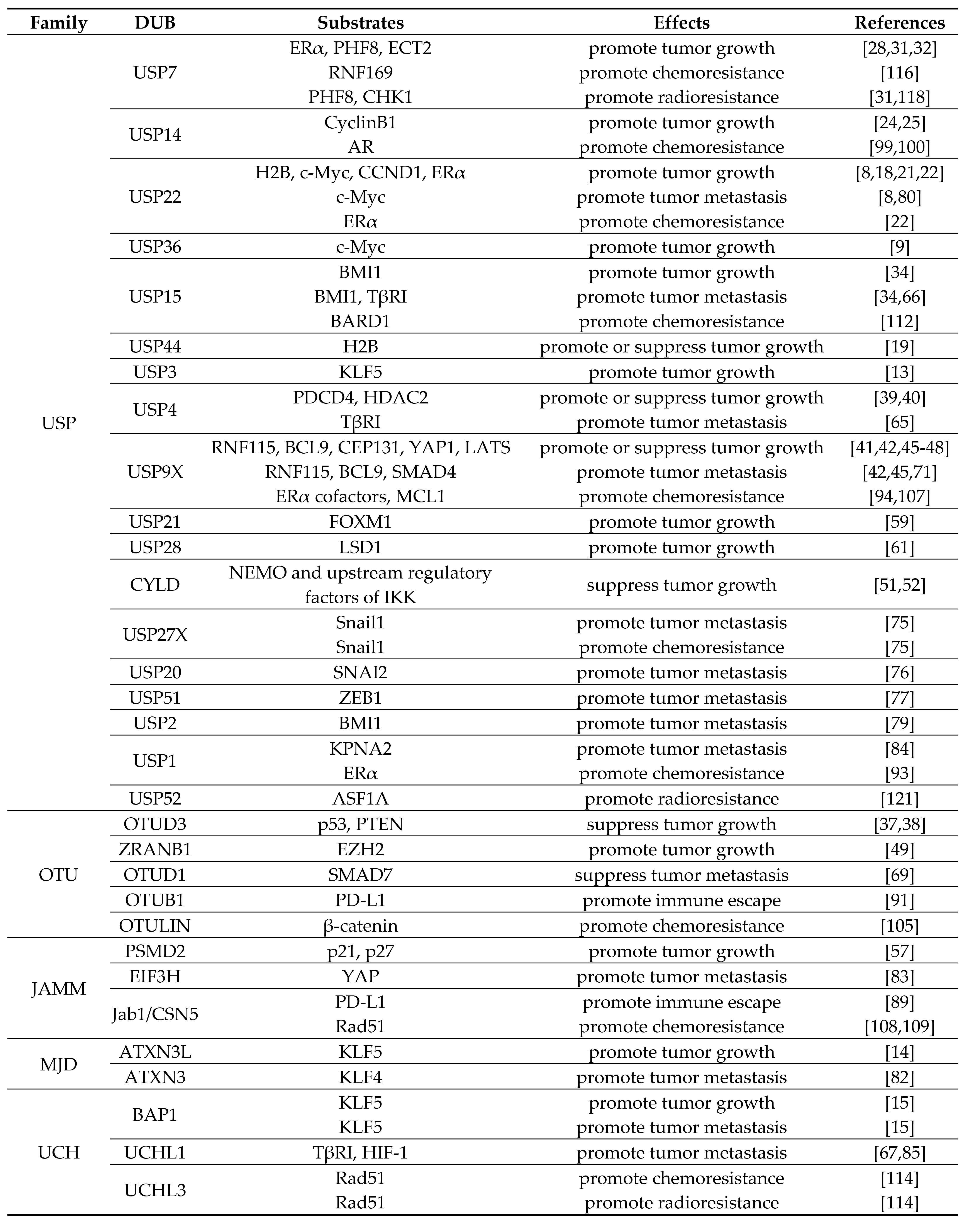
Table 1. Roles of DUBs in breast cancer progression.
References
- Lu Deng; Tong Meng; Lei Chen; Wenyi Wei; Ping Wang; The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduction and Targeted Therapy 2020, 5, 1-28, 10.1038/s41392-020-0107-0.
- Julia Martínez Fraile; Víctor Quesada; David Rodríguez; Jose M.P. Freije; Carlos López-Otín; Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 2011, 31, 2373-2388, 10.1038/onc.2011.443.
- Jeanine A. Harrigan; Xavier Jacq; Niall M. Martin; Stephen P. Jackson; Deubiquitylating enzymes and drug discovery: emerging opportunities. Nature Reviews Drug Discovery 2017, 17, 57-78, 10.1038/nrd.2017.152.
- Nadia Harbeck; Frédérique Penault-Llorca; Javier Cortes; Michael Gnant; Nehmat Houssami; Philip Poortmans; Kathryn Ruddy; Janice Tsang; Fatima Cardoso; Breast cancer. Nature Reviews Disease Primers 2019, 5, 1-31, 10.1038/s41572-019-0111-2.
- Sovana Adhikary; Martin Eilers; Transcriptional regulation and transformation by Myc proteins. Nature Reviews Molecular Cell Biology 2005, 6, 635-645, 10.1038/nrm1703.
- Liu Li; Shuo Wang; Ming Wang; Guoqing Qi; Hongliang Zhao; Ubiquitin-Specific Peptidase 5 is Involved in the Proliferation of Trophoblast Cells by Regulating Wnt/β-Catenin Signaling. Molecular Biotechnology 2021, 63, 686-693, 10.1007/s12033-021-00330-x.
- Xiao-Yong Zhang; Maya Varthi; Stephen M. Sykes; Charles Phillips; Claude Warzecha; Wenting Zhu; Anastasia Wyce; Alan W. Thorne; Shelley L. Berger; Steven B. McMahon; et al. The Putative Cancer Stem Cell Marker USP22 Is a Subunit of the Human SAGA Complex Required for Activated Transcription and Cell-Cycle Progression. Molecular Cell 2008, 29, 102-111, 10.1016/j.molcel.2007.12.015.
- Dongyeon Kim; Ahyoung Hong; Hye In Park; Woo Hyun Shin; Lang Yoo; Seo Jeong Jeon; Kwang Chul Chung; Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. Journal of Cellular Physiology 2017, 232, 3664-3676, 10.1002/jcp.25841.
- Xiao-Xin Sun; Xia He; Li Yin; Masayuki Komada; Rosalie C. Sears; Mu-Shui Dai; The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proceedings of the National Academy of Sciences 2015, 112, 3734-3739, 10.1073/pnas.1411713112.
- Mark K. Farrugia; Daniel B. Vanderbilt; Mohamad Salkeni; J. Michael Ruppert; Kruppel-like Pluripotency Factors as Modulators of Cancer Cell Therapeutic Responses. Cancer Research 2016, 76, 1677-1682, 10.1158/0008-5472.can-15-1806.
- Rong Liu; Zhongmei Zhou; Dong Zhao; Ceshi Chen; The Induction of KLF5 Transcription Factor by Progesterone Contributes to Progesterone-Induced Breast Cancer Cell Proliferation and Dedifferentiation. Molecular Endocrinology 2011, 25, 1137-1144, 10.1210/me.2010-0497.
- Dan Tong; Klaus Czerwenka; Georg Heinze; Martin Ryffel; Eva Schuster; Armin Witt; Sepp Leodolter; Robert Zeillinger; Expression of KLF5 is a Prognostic Factor for Disease-Free Survival and Overall Survival in Patients with Breast Cancer. Clinical Cancer Research 2006, 12, 2442-2448, 10.1158/1078-0432.ccr-05-0964.
- Yingying Wu; Junying Qin; Fubing Li; Chuanyu Yang; Zhen Li; Zhongmei Zhou; Hailin Zhang; Yunxi Li; Xinye Wang; Rong Liu; et al.Qian TaoWenlin ChenCeshi Chen USP3 promotes breast cancer cell proliferation by deubiquitinating KLF5. Journal of Biological Chemistry 2019, 294, 17837-17847, 10.1074/jbc.ra119.009102.
- Fei Ge; Wenlin Chen; Junying Qin; Zhongmei Zhou; Rong Liu; Linlin Liu; Jing Tan; Tianning Zou; Hongyuan Li; Guosheng Ren; et al.Ceshi Chen Ataxin-3 like (ATXN3L), a member of the Josephin family of deubiquitinating enzymes, promotes breast cancer proliferation by deubiquitinating Krüppel-like factor 5 (KLF5). Oncotarget 2015, 6, 21369-21378, 10.18632/oncotarget.4128.
- Junying Qin; Zhongmei Zhou; Wenlin Chen; Chunyan Wang; Hailin Zhang; Guangzhe Ge; Ming Shao; Dingyun You; Zhixiang Fan; HouJun Xia; et al.Rong LiuCeshi Chen BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nature Communications 2015, 6, 8471, 10.1038/ncomms9471.
- Liang Yan; Meng‐Chao Yu; Guang‐Lei Gao; Hong‐Wei Liang; Xin‐Yan Zhou; Zhouting Zhu; Chen‐Yu Zhang; Ya‐Bing Wang; Xi Chen; MiR‐125a‐5p functions as a tumour suppressor in breast cancer by downregulating BAP1. Journal of Cellular Biochemistry 2018, 119, 8773-8783, 10.1002/jcb.27124.
- Tanja Prenzel; Yvonne Begus-Nahrmann; Frank Kramer; Magali Hennion; Chieh Hsu; Theresa Gorsler; Corinna Hintermair; Dirk Eick; Elisabeth Kremmer; Mikael Simons; et al.Tim BeißbarthSteven Johnsen Estrogen-Dependent Gene Transcription in Human Breast Cancer Cells Relies upon Proteasome-Dependent Monoubiquitination of Histone H2B. Cancer Research 2011, 71, 5739-5753, 10.1158/0008-5472.can-11-1896.
- Boyko S. Atanassov; Ryan D. Mohan; Xianjiang Lan; Xianghong Kuang; Yue Lu; Kevin Lin; Elizabeth McIvor; Wenqian Li; Ying Zhang; Laurence Florens; et al.Stephanie D. ByrumSamuel G. MackintoshTammy Calhoun-DavisEvangelia KoutelouLi WangDean TangAlan J. TackettMichael WashburnJerry L. WorkmanSharon Y.R. Dent ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Molecular Cell 2016, 62, 558-571, 10.1016/j.molcel.2016.03.030.
- Ohad Tarcic; Roy Zvi Granit; Ioannis S Pateras; Hadas Masury; Bella Maly; Yaara Zwang; Yosef Yarden; Vassilis Gorgoulis; Eli Pikarsky; Ittai Ben-Porath; et al.Moshe Oren RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death & Differentiation 2017, 24, 694-704, 10.1038/cdd.2016.126.
- Matthew Ingham; Gary K. Schwartz; Cell-Cycle Therapeutics Come of Age. Journal of Clinical Oncology 2017, 35, 2949-2959, 10.1200/jco.2016.69.0032.
- Victoria J. Gennaro; Timothy J. Stanek; Amy R. Peck; Yunguang Sun; Feng Wang; Shuo Qie; Karen Knudsen; Hallgeir Rui; Tauseef Butt; J. Alan Diehl; et al.Steven B. McMahon Control of CCND1 ubiquitylation by the catalytic SAGA subunit USP22 is essential for cell cycle progression through G1 in cancer cells. Proceedings of the National Academy of Sciences 2018, 115, E9298-E9307, 10.1073/pnas.1807704115.
- Shengli Wang; Xinping Zhong; Chunyu Wang; Hao Luo; Lin Lin; Hongmiao Sun; Ge Sun; Kai Zeng; Renlong Zou; Wei Liu; et al.Ning SunHuijuan SongWensu LiuQiang ZhangZhixuan LiaoXiaochun TengTingting ZhouXun SunYue Zhao USP22 positively modulates ERα action via its deubiquitinase activity in breast cancer. Cell Death & Differentiation 2020, 27, 3131-3145, 10.1038/s41418-020-0568-2.
- Bing Liu; Jiangping Chen; Song Zhang; Emerging role of ubiquitin-specific protease 14 in oncogenesis and development of tumor: Therapeutic implication. Life Sciences 2019, 239, 116875, 10.1016/j.lfs.2019.116875.
- Lianxin Zhu; Shuyun Yang; Song He; Fulin Qiang; Jing Cai; Rong Liu; Changjiang Gu; Zengya Guo; Chen Wang; Wei Zhang; et al.Chunhui ZhangYingying Wang Downregulation of ubiquitin-specific protease 14 (USP14) inhibits breast cancer cell proliferation and metastasis, but promotes apoptosis. The Histochemical Journal 2015, 47, 69-80, 10.1007/s10735-015-9650-3.
- Bing Liu; Yuhan Liu; Yanan Wang; Caifeng Xie; Mingxi Gan; Tianyu Han; Jiaqing Cao; Jianbin Wang; CyclinB1 deubiquitination by USP14 regulates cell cycle progression in breast cancer. Pathology - Research and Practice 2019, 215, 152592, 10.1016/j.prp.2019.152592.
- Kun Ding; Wenqing Li; Zhiqiang Zou; Xianzhi Zou; Chengru Wang; CCNB1 is a prognostic biomarker for ER+ breast cancer. Medical Hypotheses 2014, 83, 359-364, 10.1016/j.mehy.2014.06.013.
- Zhiru Wang; Wenting Kang; Yinghua You; Jingru Pang; Hongmei Ren; Zhenhe Suo; Hongmin Liu; Yichao Zheng; USP7: Novel Drug Target in Cancer Therapy. Frontiers in Pharmacology 2019, 10, 427, 10.3389/fphar.2019.00427.
- Xiaohong Xia; Yuning Liao; Chuyi Huang; Yuan Liu; Jinchan He; Zhenlong Shao; Lili Jiang; Q. Ping Dou; Jinbao Liu; Hongbiao Huang; et al. Deubiquitination and stabilization of estrogen receptor α by ubiquitin-specific protease 7 promotes breast tumorigenesis. Cancer Letters 2019, 465, 118-128, 10.1016/j.canlet.2019.09.003.
- Shuyan Li; Ao Sun; Xiuming Liang; Lin Ma; Li Shen; Tongyu Li; Lixin Zheng; Wenjing Shang; Wei Zhao; Jihui Jia; et al. Histone demethylase PHF8 promotes progression and metastasis of gastric cancer. American journal of cancer research 2017, 7, 448-461.
- Qiuli Liu; Jian Pang; Lin‐Ang Wang; Zhuowei Huang; Jing Xu; Xingxia Yang; Qiubo Xie; Yiqiang Huang; Tang Tang; Dali Tong; et al.Gaolei LiuLuofu WangDianzheng ZhangQiang MaHualiang XiaoWeihua LanJun QinJun Jiang Histone demethylase PHF8 drives neuroendocrine prostate cancer progression by epigenetically upregulating FOXA2. The Journal of Pathology 2020, 253, 106-118, 10.1002/path.5557.
- Qian Wang; Shuai Ma; Nan Song; Xin Li; Ling Liu; Shangda Yang; Xiang Ding; Lin Shan; Xing Zhou; Dongxue Su; et al.Yue WangQi ZhangXinhua LiuNa YuKai ZhangYongfeng ShangZhi YaoLei Shi Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. Journal of Clinical Investigation 2016, 126, 2205-2220, 10.1172/jci85747.
- Qi Zhang; Cheng Cao; Wenchen Gong; Kaiwen Bao; Qian Wang; Yuejiao Wang; Liyuan Bi; Shuai Ma; Jiao Zhao; Ling Liu; et al.Shanshan TianKai ZhangJie YangZhi YaoNan SongLei Shi A feedforward circuit shaped by ECT2 and USP7 contributes to breast carcinogenesis. Theranostics 2020, 10, 10769-10790, 10.7150/thno.46878.
- Christophe Ginestier; Min Hee Hur; Emmanuelle Charafe-Jauffret; Florence Monville; Julie Dutcher; Marty Brown; Jocelyne Jacquemier; Patrice Viens; Celina G. Kleer; Suling Liu; et al.Anne SchottDan HayesDaniel BirnbaumMax S. WichaGabriela Dontu ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555-567, 10.1016/j.stem.2007.08.014.
- Lixing Zhang; Jiankun Qiang; Xiaoli Yang; Dong Wang; Adeel Ur Rehman; Xueyan He; Weilong Chen; Dandan Sheng; Lei Zhou; Yi‐Zhou Jiang; et al.Tao LiYing DuJing FengXin HuJian ZhangXi‐Chun HuZhi‐Ming ShaoSuling Liu IL1R2 Blockade Suppresses Breast Tumorigenesis and Progression by Impairing USP15‐Dependent BMI1 Stability. Advanced Science 2019, 7, 1901728, 10.1002/advs.201901728.
- Mathangi Srinivasan; Dhruba J. Bharali; Thangirala Sudha; Maha Khedr; Ian Guest; Stewart Sell; Gennadi V. Glinsky; Shaker A. Mousa; Downregulation of Bmi1 in breast cancer stem cells suppresses tumor growth and proliferation. Oncotarget 2017, 8, 38731-38742, 10.18632/oncotarget.16317.
- Tongde Du; Hongchang Li; Yongsheng Fan; Lin Yuan; Xiaodan Guo; Qiong Zhu; Yuying Yao; Xin Li; Chunlei Liu; Xinhe Yu; et al.Zhaofei LiuChun-Ping CuiChuanchun HanLingqiang Zhang The deubiquitylase OTUD3 stabilizes GRP78 and promotes lung tumorigenesis. Nature Communications 2019, 10, 1-15, 10.1038/s41467-019-10824-7.
- Qian Pu; Yan-Rong Lv; Ke Dong; Wen-Wen Geng; Hai-Dong Gao; Tumor suppressor OTUD3 induces growth inhibition and apoptosis by directly deubiquitinating and stabilizing p53 in invasive breast carcinoma cells. BMC Cancer 2020, 20, 1-14, 10.1186/s12885-020-07069-9.
- Lin Yuan; Yanrong Lv; Hongchang Li; Haidong Gao; Shanshan Song; Yuan Zhang; Guichun Xing; Xiangzhen Kong; Lijing Wang; Yang Li; et al.Tao ZhouDaming GaoZhi-Xiong XiaoYuxin YinWenyi WeiFuchu HeLingqiang Zhang Deubiquitylase OTUD3 regulates PTEN stability and suppresses tumorigenesis. Nature 2015, 17, 1169-1181, 10.1038/ncb3218.
- Yang Li; Daqing Jiang; Qi Zhang; Xiaoli Liu; Zhengang Cai; Ubiquitin-specific protease 4 inhibits breast cancer cell growth through the upregulation of PDCD4. International Journal of Molecular Medicine 2016, 38, 803-811, 10.3892/ijmm.2016.2685.
- Yuzhi Wang; Jun Zhang; Lele Wu; Weiguang Liu; Guanyun Wei; Xue Gong; Yan Liu; Zhifang Ma; Fei Ma; Jean Paul Thiery; et al.Liming Chen Tricho-rhino-phalangeal syndrome 1 protein functions as a scaffold required for ubiquitin-specific protease 4-directed histone deacetylase 2 de-ubiquitination and tumor growth. Breast Cancer Research 2018, 20, 83, 10.1186/s13058-018-1018-7.
- Hang Li; Bin Zheng; Overexpression of the Ubiquitin-Specific Peptidase 9 X-Linked (USP9X) Gene is Associated with Upregulation of Cyclin D1 (CCND1) and Downregulation of Cyclin-Dependent Inhibitor Kinase 1A (CDKN1A) in Breast Cancer Tissue and Cell Lines. Medical Science Monitor 2019, 25, 4207-4216, 10.12659/msm.914742.
- Qin Lu; Dayun Lu; Zhi-Ming Shao; Da-Qiang Li; Deubiquitinase ubiquitin‐specific protease 9X regulates the stability and function of E3 ubiquitin ligase ring finger protein 115 in breast cancer cells. Cancer Science 2019, 110, 1268-1278, 10.1111/cas.13953.
- Zehua Wang; Zhi Nie; Wenlin Chen; Zhongmei Zhou; Qinghua Kong; Arun K. Seth; Rong Liu; Ceshi Chen; RNF115/BCA2 E3 Ubiquitin Ligase Promotes Breast Cancer Cell Proliferation through Targeting p21Waf1/Cip1 for Ubiquitin-Mediated Degradation. Neoplasia 2013, 15, 1028-1035, 10.1593/neo.13678.
- Alexandra Klaus-Bergmann; Walter Birchmeier; Wnt signalling and its impact on development and cancer. Nature Reviews Cancer 2008, 8, 387-398, 10.1038/nrc2389.
- Zesen Shang; Jiao Zhao; Qi Zhang; Cheng Cao; Shanshan Tian; Kai Zhang; Ling Liu; Lei Shi; Na Yu; Shangda Yang; et al. USP9X-mediated deubiquitination of B-cell CLL/lymphoma 9 potentiates Wnt signaling and promotes breast carcinogenesis. Journal of Biological Chemistry 2019, 294, 9844-9857, 10.1074/jbc.ra119.007655.
- Xin Li; Nan Song; Ling Liu; Xinhua Liu; Xiang Ding; Xin Song; Shangda Yang; Lin Shan; Xing Zhou; Dongxue Su; et al.Yue WangQi ZhangCheng CaoShuai MaNa YuFuquan YangYan WangZhi YaoYongfeng ShangLei Shi USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nature Communications 2017, 8, 14866, 10.1038/ncomms14866.
- Lei Li; Tongzheng Liu; Yunhui Li; Chenming Wu; Kuntian Luo; Yujiao Yin; Yuping Chen; Somaira Nowsheen; Jinhuan Wu; Zhenkun Lou; et al.Jian Yuan The deubiquitinase USP9X promotes tumor cell survival and confers chemoresistance through YAP1 stabilization. Oncogene 2018, 37, 2422-2431, 10.1038/s41388-018-0134-2.
- Aleksandra Toloczko; Fusheng Guo; Hiu-Fung Yuen; Qing Wen; Stephen Wood; Yan Shan Ong; Pei Yi Chan; Asfa Alli-Shaik; Jayantha Gunaratne; Mark J. Dunne; et al.Wanjin HongSiew Wee Chan Deubiquitinating Enzyme USP9X Suppresses Tumor Growth via LATS Kinase and Core Components of the Hippo Pathway. Cancer Research 2017, 77, 4921-4933, 10.1158/0008-5472.can-16-3413.
- Peijing Zhang; Zhenna Xiao; Shouyu Wang; Mutian Zhang; Yongkun Wei; Qinglei Hang; Jongchan Kim; Fan Yao; Cristian Rodriguez-Aguayo; Baochau N. Ton; et al.Minjung LeeYumeng WangZhicheng ZhouLiyong ZengXiaoyu HuSarah E. LawhonAshley N. SiverlyXiaohua SuJia LiXiaoping XieXuhong ChengLiang-Chiu LiuHui-Wen ChangShu-Fen ChiangGabriel Lopez-BeresteinAnil K. SoodJunjie ChenM. James YouShao-Cong SunHan LiangYun HuangXianbin YangDeqiang SunYutong SunMien-Chie HungLi Ma ZRANB1 Is an EZH2 Deubiquitinase and a Potential Therapeutic Target in Breast Cancer. Cell Reports 2018, 23, 823-837, 10.1016/j.celrep.2018.03.078.
- S-C Sun; CYLD: a tumor suppressor deubiquitinase regulating NF-κB activation and diverse biological processes. Cell Death & Differentiation 2009, 17, 25-34, 10.1038/cdd.2009.43.
- Mitsuhiro Hayashi; Hirofumi Jono; Satoru Shinriki; Takuya Nakamura; Jianying Guo; Aiko Sueta; Mai Tomiguchi; Saori Fujiwara; Mutsuko Yamamoto-Ibusuki; Kei-Ichi Murakami; et al.Satoshi YamashitaYutaka YamamotoJian-Dong LiHirotaka IwaseYukio Ando Clinical significance of CYLD downregulation in breast cancer. Breast Cancer Research and Treatment 2014, 143, 447-457, 10.1007/s10549-013-2824-3.
- Timoklia Orfanidou; Konstantinos Xanthopoulos; Dimitra Dafou; Athanasios Pseftogas; Paul Hadweh; Claire Psyllaki; Eudoxia Hatzivassiliou; George Mosialos; Down-regulation of the Tumor Suppressor CYLD Enhances the Transformed Phenotype of Human Breast Cancer Cells. Anticancer Research 2017, 37, 3493-3503, 10.21873/anticanres.11717.
- Thijn R. Brummelkamp; Sebastian M. B. Nijman; Annette M. G. Dirac; Rene Bernards; Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 2003, 424, 797-801, 10.1038/nature01811.
- Zhaojun Ren; Mengmeng Lv; Qiao Yu; Jun Bao; Kexin Lou; Xiujuan Li; MicroRNA‐370‐3p shuttled by breast cancer cell‐derived extracellular vesicles induces fibroblast activation through the CYLD/Nf‐κB axis to promote breast cancer progression. The FASEB Journal 2021, 35, e21383, 10.1096/fj.202001430rr.
- Hongming Song; Dengfeng Li; Tianqi Wu; Dan Xie; Kaiyao Hua; Jiashu Hu; Xiaochong Deng; Changle Ji; Yijun Deng; Lin Fang; et al. MicroRNA-301b promotes cell proliferation and apoptosis resistance in triple-negative breast cancer by targeting CYLD. BMB Reports 2018, 51, 602-607, 10.5483/bmbrep.2018.51.11.168.
- Yasushi Matsuyama; Motoshi Suzuki; Chinatsu Arima; Qin Miao Huang; Shuta Tomida; Toshiyuki Takeuchi; Ryoji Sugiyama; Yasutomo Itoh; Yasushi Yatabe; Hidemi Goto; et al.Takashi Takahashi Proteasomal non-catalytic subunit PSMD2 as a potential therapeutic target in association with various clinicopathologic features in lung adenocarcinomas. Molecular Carcinogenesis 2011, 50, 301-309, 10.1002/mc.20632.
- Yunhai Li; Jing Huang; Beilei Zeng; Dejuan Yang; Jiazheng Sun; Xuedong Yin; Mengqi Lu; Zhu Qiu; Weiyan Peng; Tingxiu Xiang; et al.Hongzhong LiGuosheng Ren PSMD2 regulates breast cancer cell proliferation and cell cycle progression by modulating p21 and p27 proteasomal degradation. Cancer Letters 2018, 430, 109-122, 10.1016/j.canlet.2018.05.018.
- Liang Peng; Yi Hu; Demeng Chen; Shunchang Jiao; Shengkun Sun; Ubiquitin specific peptidase 21 regulates interleukin-8 expression, stem-cell like property of human renal cell carcinoma. Oncotarget 2016, 7, 42007-42016, 10.18632/oncotarget.9751.
- Anthony Arceci; Thomas Bonacci; Xianxi Wang; Kyle Stewart; Jeffrey S. Damrauer; Katherine Hoadley; Michael J. Emanuele; FOXM1 Deubiquitination by USP21 Regulates Cell Cycle Progression and Paclitaxel Sensitivity in Basal-like Breast Cancer. Cell Reports 2019, 26, 3076-3086.e6, 10.1016/j.celrep.2019.02.054.
- Xiaofang Wang; Zhiyi Liu; Li Zhang; Zhaozhi Yang; Xingxing Chen; Jurui Luo; Zhirui Zhou; Xin Mei; Xiaoli Yu; Zhimin Shao; et al.Yan FengShen FuZhen ZhangDongping WeiLijun JiaJinli MaXiaomao Guo Targeting deubiquitinase USP28 for cancer therapy. Cell Death & Disease 2018, 9, 1-10, 10.1038/s41419-017-0208-z.
- Yadi Wu; Yifan Wang; Xiuwei H. Yang; Tiebang Kang; Yongxiang Zhao; Chi Wang; B. Mark Evers; Binhua P. Zhou; The Deubiquitinase USP28 Stabilizes LSD1 and Confers Stem-Cell-like Traits to Breast Cancer Cells. Cell Reports 2013, 5, 224-236, 10.1016/j.celrep.2013.08.030.
- Warren A. Whyte; Steve Bilodeau; David A. Orlando; Heather A. Hoke; Garrett Frampton; Charles T. Foster; Shaun Cowley; Richard A. Young; Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 2012, 482, 221-225, 10.1038/nature10805.
- Chunyu Cao; S N Vasilatos; R Bhargava; J L Fine; S Oesterreich; N E Davidson; Yi Huang; Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 2016, 36, 133-145, 10.1038/onc.2016.186.
- Cindy Neuzillet; Annemilaï Tijeras-Raballand; Romain Cohen; Jerome Cros; Sandrine Faivre; Eric Raymond; Armand de Gramont; Targeting the TGFβ pathway for cancer therapy. Pharmacology & Therapeutics 2015, 147, 22-31, 10.1016/j.pharmthera.2014.11.001.
- Long Zhang; Fangfang Zhou; Yvette Drabsch; Rui Gao; B. Ewa Snaar-Jagalska; Craig Mickanin; Huizhe Huang; Kelly-Ann Sheppard; Jeff A. Porter; Chris X. Lu; et al.Peter Ten Dijke USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nature 2012, 14, 717-726, 10.1038/ncb2522.
- Pieter Eichhorn; Laura Rodón; Alba Gonzàlez-Juncà; Annette Dirac; Magüi Gili; Elena Martinez-Saez; Claudia Aura; Ignasi Barba; Vicente Peg; Aleix Prat; et al.Isabel CuartasJose JimenezDavid García-DoradoJuan SahuquilloRene BernardsJosé BaselgaJoan Seoane USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nature Medicine 2012, 18, 429-435, 10.1038/nm.2619.
- Sijia Liu; Román González-Prieto; Mengdi Zhang; Paul P. Geurink; Raymond Kooij; Prasanna Vasudevan Iyengar; Maarten van Dinther; Erik Bos; Xiaobing Zhang; Sylvia E. Le Dévédec; et al.Bob van de WaterRoman I. KoningHong-Jian ZhuWilma E. MeskerAlfred C.O. VertegaalHuib OvaaLong ZhangJohn W.M. MartensPeter Ten Dijke Deubiquitinase Activity Profiling Identifies UCHL1 as a Candidate Oncoprotein That Promotes TGFβ-Induced Breast Cancer Metastasis. Clinical Cancer Research 2019, 26, 1460-1473, 10.1158/1078-0432.ccr-19-1373.
- Long Zhang; Fangfang Zhou; Amaya García de Vinuesa; Esther M. de Kruijf; Wilma E. Mesker; Li Hui; Yvette Drabsch; Yihao Li; Andreas Bauer; Adrien Rousseau; et al.Kelly-Ann SheppardCraig MickaninPeter KuppenChris X. LuPeter Ten Dijke TRAF4 Promotes TGF-β Receptor Signaling and Drives Breast Cancer Metastasis. Molecular Cell 2013, 51, 559-572, 10.1016/j.molcel.2013.07.014.
- Zhengkui Zhang; Yao Fan; Feng Xie; Hang Zhou; Ke Jin; Li Shao; Wenhao Shi; Pengfei Fang; Bing Yang; Hans van Dam; et al.Peter Ten DijkeXiaofeng ZhengXiaohua YanJunling JiaMin ZhengJin JinChen DingSheng YeFangfang ZhouLong Zhang Breast cancer metastasis suppressor OTUD1 deubiquitinates SMAD7. Nature Communications 2017, 8, 1-16, 10.1038/s41467-017-02029-7.
- Peter Kavsak; Richele K. Rasmussen; Carrie G. Causing; Shirin Bonni; Haitao Zhu; Gerald H. Thomsen; Jeffrey L. Wrana; Smad7 Binds to Smurf2 to Form an E3 Ubiquitin Ligase that Targets the TGFβ Receptor for Degradation. Molecular Cell 2000, 6, 1365-1375, 10.1016/s1097-2765(00)00134-9.
- Yong Wu; Xiaoting Yu; Xianghua Yi; Ke Wu; Sami Dwabe; Mohammad Atefi; Yahya Elshimali; Kevin T. Kemp; Kruttika Bhat; Jesse Haro; et al.Marianna SarkissyanJaydutt V. Vadgama Aberrant Phosphorylation of SMAD4 Thr277-Mediated USP9x-SMAD4 Interaction by Free Fatty Acids Promotes Breast Cancer Metastasis.. Cancer Research 2017, 77, 1383-1394, 10.1158/0008-5472.CAN-16-2012.
- Samy Lamouille; Jian Xu; Rik Derynck; Molecular mechanisms of epithelial–mesenchymal transition. Nature Reviews Molecular Cell Biology 2014, 15, 178-196, 10.1038/nrm3758.
- Vivek Mittal; Epithelial Mesenchymal Transition in Tumor Metastasis. Annual Review of Pathology: Mechanisms of Disease 2018, 13, 395-412, 10.1146/annurev-pathol-020117-043854.
- Yifan Wang; Jian Shi; Kequn Chai; Xuhua Ying; Binhua P. Zhou; The Role of Snail in EMT and Tumorigenesis. Current Cancer Drug Targets 2013, 13, 963-972, 10.2174/15680096113136660102.
- Guillem Lambies; Martina Miceli; Catalina Martínez-Guillamon; Rubén Olivera-Salguero; Raúl Peña; Carolina-Paola Frías; Irene Calderón; Boyko Atanassov; Sharon Y. R. Dent; Joaquin Arribas; et al.Antonio García De HerrerosVíctor M. Díaz TGFβ-Activated USP27X Deubiquitinase Regulates Cell Migration and Chemoresistance via Stabilization of Snail1. Cancer Research 2018, 79, 33-46, 10.1158/0008-5472.can-18-0753.
- Wenyang Li; Minhong Shen; Yi-Zhou Jiang; Ruina Zhang; Hanqiu Zheng; Yong Wei; Zhi-Ming Shao; Yibin Kang; Deubiquitinase USP20 promotes breast cancer metastasis by stabilizing SNAI2. Genes & Development 2020, 34, 1310-1315, 10.1101/gad.339804.120.
- Zhen Zhang; Jianjun Li; Yang Ou; Guang Yang; KaiYuan Deng; Qiong Wang; Zhaoyang Wang; Wenhao Wang; Quansheng Zhang; Hang Wang; et al.Wei SunPeiqing SunShuang Yang CDK4/6 inhibition blocks cancer metastasis through a USP51-ZEB1-dependent deubiquitination mechanism. Signal Transduction and Targeted Therapy 2020, 5, 25, 10.1038/s41392-020-0118-x.
- Li-Bing Song; Jun Li; Wen-Ting Liao; Yan Feng; Chun-Ping Yu; Li-Juan Hu; Qing-Li Kong; Li-Hua Xu; Xing Zhang; Wan-Li Liu; et al.Man-Zhi LiTie-Bang KangLi-Wu FuWen-Lin HuangYun-Fei XiaSai Wah TsaoMengfeng LiVimla BandHamid BandQing-Hua ShiYi-Xin ZengMusheng Zeng The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. Journal of Clinical Investigation 2009, 119, 3626-3636, 10.1172/jci39374.
- Jiabei He; Hong-Jen Lee; Suchandrima Saha; Diane Ruan; Hua Guo; Chia-Hsin Chan; Inhibition of USP2 eliminates cancer stem cells and enhances TNBC responsiveness to chemotherapy. Cell Death & Disease 2019, 10, 1-16, 10.1038/s41419-019-1512-6.
- Youxue Zhang; Lei Yao; Xianyu Zhang; Hongfei Ji; Lihong Wang; Shanshan Sun; Da Pang; Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. Journal of Cancer Research and Clinical Oncology 2011, 137, 1245-1253, 10.1007/s00432-011-0998-9.
- Xiaoting Jia; Lejuan Shi; Xiaorong Wang; Liyun Luo; Li Ling; Jiang Yin; Ying Song; Zhijie Zhang; Ni Qiu; Hao Liu; et al.Min DengZhimin HeHongsheng LiGuopei Zheng KLF5 regulated lncRNA RP1 promotes the growth and metastasis of breast cancer via repressing p27kip1 translation. Cell Death & Disease 2019, 10, 373, 10.1038/s41419-019-1566-5.
- Haojing Zou; Hongyan Chen; Zhuan Zhou; Yong Wan; Zhihua Liu; ATXN3 promotes breast cancer metastasis by deubiquitinating KLF4. Cancer Letters 2019, 467, 19-28, 10.1016/j.canlet.2019.09.012.
- Zhuan Zhou; Honghong Zhou; Luca Ponzoni; Aiping Luo; Rui Zhu; Mingjing He; Yi Huang; Kun-Liang Guan; Ivet Bahar; Zhihua Liu; et al.Yong Wan EIF3H Orchestrates Hippo Pathway–Mediated Oncogenesis via Catalytic Control of YAP Stability. Cancer Research 2020, 80, 2550-2563, 10.1158/0008-5472.can-19-3718.
- Aihui Ma; Ming Tang; Li Zhang; Boshi Wang; Zhaojuan Yang; Yun Liu; Guiqin Xu; Lin Wu; Tiantian Jing; Xiaoli Xu; et al.Shengli YangYongzhong Liu USP1 inhibition destabilizes KPNA2 and suppresses breast cancer metastasis. Oncogene 2018, 38, 2405-2419, 10.1038/s41388-018-0590-8.
- Yoko Goto; Lihua Zeng; Chan Joo Yeom; Yuxi Zhu; Akiyo Morinibu; Kazumi Shinomiya; Minoru Kobayashi; Kiichi Hirota; Satoshi Itasaka; Michio Yoshimura; et al.Keiji TanimotoMasae ToriiTerumasa SowaToshi MenjuMakoto SonobeHideaki KakeyaMasakazu ToiHiroshi DateEster M. HammondMasahiro HiraokaHiroshi Harada UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1α. Nature Communications 2015, 6, 6153, 10.1038/ncomms7153.
- H Zhang; Carmen Chak Lui Wong; H Wei; D M Gilkes; Preethi Korangath; P Chaturvedi; L Schito; Jasper Chen; B Krishnamachary; P T Winnard; et al.V RamanL ZhenW A MitznerS SukumarG L Semenza HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2011, 31, 1757-1770, 10.1038/onc.2011.365.
- Mary E. Keir; Manish J. Butte; Gordon J. Freeman; Arlene H. Sharpe; PD-1 and Its Ligands in Tolerance and Immunity. Annual Review of Immunology 2008, 26, 677-704, 10.1146/annurev.immunol.26.021607.090331.
- FeiTing Xie; Mengxue Xu; Jian Lu; Lingxiang Mao; Shengjun Wang; The role of exosomal PD-L1 in tumor progression and immunotherapy. Molecular Cancer 2019, 18, 1-10, 10.1186/s12943-019-1074-3.
- Seung-Oe Lim; Chia-Wei Li; Weiya Xia; Jong-Ho Cha; Li-Chuan Chan; Yun Wu; Shih-Shin Chang; Wan-Chi Lin; Jung-Mao Hsu; Yi-Hsin Hsu; et al.Taewan KimWei-Chao ChangJennifer L. HsuHirohito YamaguchiQingqing DingYan WangYi YangChung-Hsuan ChenAysegul A. SahinDihua YuGabriel N. HortobagyiMien-Chie Hung Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925-939, 10.1016/j.ccell.2016.10.010.
- Ming Zhang; Ning Wang; Peng Song; Yingqiang Fu; Yanlv Ren; Zhigao Li; Jinsong Wang; LncRNA GATA3‐AS1 facilitates tumour progression and immune escape in triple‐negative breast cancer through destabilization of GATA3 but stabilization of PD‐L1. Cell Proliferation 2020, 53, e12855, 10.1111/cpr.12855.
- Dan Zhu; Ruidan Xu; Xinping Huang; Zefang Tang; Yonglu Tian; Jinfang Zhang; Xiaofeng Zheng; Deubiquitinating enzyme OTUB1 promotes cancer cell immunosuppression via preventing ER-associated degradation of immune checkpoint protein PD-L1. Cell Death & Differentiation 2020, 28, 1773-1789, 10.1038/s41418-020-00700-z.
- Simak Ali; R. Charles Coombes; Endocrine-responsive breast cancer and strategies for combating resistance. Nature Cancer 2002, 2, 101-112, 10.1038/nrc721.
- Zhiguo Niu; Xin Li; Suyin Feng; Qingsong Huang; Ting Zhuang; Cheng Yan; Hui Qian; Yinlu Ding; Jian Zhu; Wenrong Xu; et al. The deubiquitinating enzyme USP1 modulates ERα and modulates breast cancer progression. Journal of Cancer 2020, 11, 6992-7000, 10.7150/jca.50477.
- Hendrika M. Oosterkamp; Marielle Hijmans; Thijn R. Brummelkamp; Sander Canisius; Lodewyk F.A. Wessels; Wilbert Zwart; René Bernards; USP9X Downregulation Renders Breast Cancer Cells Resistant to Tamoxifen. Cancer Research 2014, 74, 3810-3820, 10.1158/0008-5472.can-13-1960.
- Chad J. Creighton; Amy M. Hilger; Shalini Murthy; James M. Rae; Arul M. Chinnaiyan; Dorraya El-Ashry; Activation of Mitogen-Activated Protein Kinase in Estrogen Receptor α–Positive Breast Cancer Cells In vitro Induces an In vivo Molecular Phenotype of Estrogen Receptor α–Negative Human Breast Tumors. Cancer Research 2006, 66, 3903-3911, 10.1158/0008-5472.can-05-4363.
- Xi-Sha Chen; Kuan-Song Wang; Wei Guo; Lan-Ya Li; Pian Yu; Xin-Yuan Sun; Hai-Yan Wang; Yi-Di Guan; Yong-Guang Tao; Bo-Ni Ding; et al.Ming-Zhu YinXing-Cong RenYi ZhangCe-Shi ChenYuan-Chao YeJin-Ming YangYan Cheng UCH-L1-mediated Down-regulation of Estrogen Receptor α Contributes to Insensitivity to Endocrine Therapy for Breast Cancer. Theranostics 2020, 10, 1833-1848, 10.7150/thno.39814.
- Laura C Collins; Kimberly S Cole; Jonathan D Marotti; Rong Hu; Stuart J Schnitt; Rulla M Tamimi; Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Modern Pathology 2011, 24, 924-931, 10.1038/modpathol.2011.54.
- Miho Kono; Takeo Fujii; Bora Lim; Meghan Sri Karuturi; Debasish Tripathy; Naoto T. Ueno; Androgen Receptor Function and Androgen Receptor–Targeted Therapies in Breast Cancer. JAMA Oncology 2017, 3, 1266-1273, 10.1001/jamaoncol.2016.4975.
- Yuning Liao; Xiaohong Xia; Ningning Liu; Jianyu Cai; Zhiqiang Guo; Yanling Li; Lili Jiang; Q. Ping Dou; Daolin Tang; Hongbiao Huang; et al.Jinbao Liu Growth arrest and apoptosis induction in androgen receptor-positive human breast cancer cells by inhibition of USP14-mediated androgen receptor deubiquitination. Oncogene 2018, 37, 1896-1910, 10.1038/s41388-017-0069-z.
- Xiaohong Xia; Chuyi Huang; Yuning Liao; Yuan Liu; Jinchan He; Zhiqiang Guo; Lili Jiang; Xuejun Wang; Jinbao Liu; Hongbiao Huang; et al. Inhibition of USP14 enhances the sensitivity of breast cancer to enzalutamide. Journal of Experimental & Clinical Cancer Research 2019, 38, 1-17, 10.1186/s13046-019-1227-7.
- Jinfeng Shi; Jingjing Li; Jiaxin Li; Renkai Li; Xiaoping Wu; Fei Gao; Liang Zou; Winston Wing Shum Mak; Chaomei Fu; Jinming Zhang; et al.George Pak-Heng Leung Synergistic breast cancer suppression efficacy of doxorubicin by combination with glycyrrhetinic acid as an angiogenesis inhibitor. Phytomedicine 2020, 81, 153408, 10.1016/j.phymed.2020.153408.
- Michelle E. Melisko; Michael Assefa; Jimmy Hwang; Amy DeLuca; John W. Park; Hope S. Rugo; Phase II study of irinotecan and temozolomide in breast cancer patients with progressing central nervous system disease. Breast Cancer Research and Treatment 2019, 177, 401-408, 10.1007/s10549-019-05309-6.
- Haitao Wang; Sen Guo; Seung-Jin Kim; Fangyuan Shao; Joshua Wing Kei Ho; Kuan Un Wong; Zhengqiang Miao; Dapeng Hao; Ming Zhao; Jun Xu; et al.Jianming ZengKoon Ho WongLijun DiAda Hang-Heng WongXiaoling XuChu-Xia Deng Cisplatin prevents breast cancer metastasis through blocking early EMT and retards cancer growth together with paclitaxel. Theranostics 2021, 11, 2442-2459, 10.7150/thno.46460.
- Klaus Heger; Katherine E. Wickliffe; Ada Ndoja; Juan Zhang; Aditya Murthy; Debra L. Dugger; Allie Maltzman; Felipe De Sousa E Melo; Jeffrey Hung; Yi Zeng; et al.Erik VerschuerenDonald KirkpatrickDomagoj VucicWyne P. LeeMerone Roose-GirmaRobert J. NewmanSoren WarmingYi-Chun HsiaoLászló G. KőművesJoshua D. WebsterKim NewtonVishva M. Dixit OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 2018, 559, 120-124, 10.1038/s41586-018-0256-2.
- Wei Wang; Mingqi Li; Suriyan Ponnusamy; Yayun Chi; Jingyan Xue; Beshoy Fahmy; Meiyun Fan; Gustavo A. Miranda-Carboni; Ramesh Narayanan; Jiong Wu; et al.Zhao-Hui Wu ABL1-dependent OTULIN phosphorylation promotes genotoxic Wnt/β-catenin activation to enhance drug resistance in breast cancers. Nature Communications 2020, 11, 1-15, 10.1038/s41467-020-17770-9.
- PeiFen Fu; Feiya Du; Yu Liu; Minya Yao; Shufeng Zhang; Xiaoxiao Zheng; ShuSen Zheng; WP1130 increases cisplatin sensitivity through inhibition of usp9x in estrogen receptor-negative breast cancer cells. American journal of translational research 2017, 9, 1783-1791.
- Martin Schwickart; Xiaodong Huang; Jennie R. Lill; Jinfeng Liu; Ronald Ferrando; Dorothy M. French; Heather Maecker; Karen O’Rourke; Fernando Bazan; Jeffrey Eastham-Anderson; et al.Peng YueDavid DornanDavid HuangVishva M. Dixit Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2009, 463, 103-107, 10.1038/nature08646.
- Sumei Wang; Do-Youn Oh; Vasiliki Leventaki; Elias Drakos; Ronghua Zhang; Aysegul A. Sahin; Erika Resetkova; Mary Elizabeth Edgerton; Wanyin Wu; Francois X. Claret; et al. MicroRNA-17 acts as a tumor chemosensitizer by targeting JAB1/CSN5 in triple-negative breast cancer. Cancer Letters 2019, 465, 12-23, 10.1016/j.canlet.2019.08.016.
- Guohong Liu; Mingxia Yu; Balu Wu; Shuang Guo; Xin Huang; Fuling Zhou; Francois X. Claret; Yunbao Pan; Jab1/Cops5 contributes to chemoresistance in breast cancer by regulating Rad51. Cellular Signalling 2018, 53, 39-48, 10.1016/j.cellsig.2018.09.010.
- Nawneet K. Kurrey; Swati P. Jalgaonkar; Alok V. Joglekar; Avinash D. Ghanate; Prasad D. Chaskar; Rahul Y. Doiphode; Sharmila A. Bapat; Snail and Slug Mediate Radioresistance and Chemoresistance by Antagonizing p53-Mediated Apoptosis and Acquiring a Stem-Like Phenotype in Ovarian Cancer Cells. STEM CELLS 2009, 27, 2059-2068, 10.1002/stem.154.
- Hannah Farmer; Nuala McCabe; Christopher Lord; Andrew N. J. Tutt; Damian A. Johnson; Tobias B. Richardson; Manuela Santarosa; Krystyna J. Dillon; Ian Hickson; Charlotte Knights; et al.Niall M. B. MartinStephen Philip JacksonGraeme C. M. SmithAlan Ashworth Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917-921, 10.1038/nature03445.
- Yihan Peng; Qingchao Liao; Wei Tan; Changmin Peng; Zhaohua Hu; Yali Chen; Zhuqing Li; Jing Li; Bei Zhen; Wenge Zhu; et al.Xiangpan LiYi YaoQibin SongChengsheng LiuXiangdong QiFuchu HeHuadong Pei The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nature Communications 2019, 10, 1-15, 10.1038/s41467-019-09232-8.
- Joseph San Filippo; Patrick Sung; Hannah Klein; Mechanism of Eukaryotic Homologous Recombination. Annual Review of Biochemistry 2008, 77, 229-257, 10.1146/annurev.biochem.77.061306.125255.
- Kuntian Luo; Lei Li; Yunhui Li; Chenming Wu; Yujiao Yin; Yuping Chen; Min Deng; Somaira Nowsheen; Jian Yuan; Zhenkun Lou; et al. A phosphorylation–deubiquitination cascade regulates the BRCA2–RAD51 axis in homologous recombination. Genes & Development 2016, 30, 2581-2595, 10.1101/gad.289439.116.
- Maria Poulsen; Claudia Lukas; Jiri Lukas; Simon Bekker-Jensen; Niels Mailand; Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. Journal of Cell Biology 2012, 197, 189-199, 10.1083/jcb.201109100.
- Liwei An; Yiyang Jiang; Howin H. W. Ng; Ellen P. S. Man; Jie Chen; Ui-Soon Khoo; Qingguo Gong; Michael S. Y. Huen; Dual-utility NLS drives RNF169-dependent DNA damage responses. Proceedings of the National Academy of Sciences 2017, 114, E2872-E2881, 10.1073/pnas.1616602114.
- Shideng Bao; Qiulian Wu; Roger E. McLendon; Yueling Hao; Qing Shi; Anita B. Hjelmeland; Mark W. Dewhirst; Darell D. Bigner; Jeremy N. Rich; Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756-760, 10.1038/nature05236.
- Peijing Zhang; Yongkun Wei; Li Wang; Bisrat G. Debeb; Yuan Yuan; Jinsong Zhang; Jingsong Yuan; Min Wang; Dahu Chen; Yutong Sun; et al.Wendy WoodwardYongqing LiuDouglas C. DeanHan LiangTony HuK. Kian AngMien-Chie HungJunjie ChenLi Ma ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nature Cell Biology 2014, 16, 864-875, 10.1038/ncb3013.
- Baiyao Wang; Jieling Zheng; Rong Li; Yunhong Tian; Jie Lin; Yingying Liang; Quanquan Sun; Anan Xu; Ronghui Zheng; Mengzhong Liu; et al.Aimin JiJunguo BuYawei Yuan Long noncoding RNA LINC02582 acts downstream of miR-200c to promote radioresistance through CHK1 in breast cancer cells. Cell Death & Disease 2019, 10, 1-15, 10.1038/s41419-019-1996-0.
- Harry O. King; Tim Brend; Helen L. Payne; Alexander Wright; Thomas Ward; Karan Patel; Teklu Egnuni; Lucy F. Stead; Anjana Patel; Heiko Wurdak; et al.Susan C. Short RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Reports 2017, 8, 125-139, 10.1016/j.stemcr.2016.12.005.
- Shangda Yang; Ling Liu; Cheng Cao; Nan Song; Yuejiao Wang; Shuai Ma; Qi Zhang; Na Yu; Xiang Ding; Fuquan Yang; et al.Shanshan TianKai ZhangTao SunJie YangZhi YaoShaoyuan WuLei Shi USP52 acts as a deubiquitinase and promotes histone chaperone ASF1A stabilization. Nature Communications 2018, 9, 1-17, 10.1038/s41467-018-03588-z.
- Ryota Nakashima; Yoko Goto; Sho Koyasu; Minoru Kobayashi; Akiyo Morinibu; Michio Yoshimura; Masahiro Hiraoka; Ester M. Hammond; Hiroshi Harada; UCHL1-HIF-1 axis-mediated antioxidant property of cancer cells as a therapeutic target for radiosensitization. Scientific Reports 2017, 7, 6879, 10.1038/s41598-017-06605-1.

