Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dmitry Karpov | + 3620 word(s) | 3620 | 2021-11-03 10:20:19 | | | |
| 2 | Catherine Yang | Meta information modification | 3620 | 2022-03-22 09:19:10 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Karpov, D. Development of Vibrio cholerae Candidate Vaccine Strain. Encyclopedia. Available online: https://encyclopedia.pub/entry/15691 (accessed on 25 July 2026).
Karpov D. Development of Vibrio cholerae Candidate Vaccine Strain. Encyclopedia. Available at: https://encyclopedia.pub/entry/15691. Accessed July 25, 2026.
Karpov, Dmitry. "Development of Vibrio cholerae Candidate Vaccine Strain" Encyclopedia, https://encyclopedia.pub/entry/15691 (accessed July 25, 2026).
Karpov, D. (2021, November 03). Development of Vibrio cholerae Candidate Vaccine Strain. In Encyclopedia. https://encyclopedia.pub/entry/15691
Karpov, Dmitry. "Development of Vibrio cholerae Candidate Vaccine Strain." Encyclopedia. Web. 03 November, 2021.
Copy Citation
Approximately 1/6 of humanity is at high risk of experiencing cholera epidemics. Therefore, сholera epidemics and outbreaks represent one of the global health threats. Vaccination is an effective global health measure to prevent cholera epidemics. There is a need for the development of more effective, stable, and safe vaccines against cholera. We describe a strategy to genetically engineer a nontoxigenic natural Vibrio cholerae strain to develop a genetically stable and relatively safe candidate vaccine strain. Our approach is applicable for developing vaccine strains against cholera and other important human pathogens.
Vibrio cholerae
genome engineering
synthetic reporter operon
amilCP
candidate vaccine strain
1. Introduction
Pathogenic strains of Vibrio cholerae cause cholera, an acute infectious disease characterized by a sharp loss of water due to profuse diarrhoea, which can be fatal in the absence of rehydration therapy [1]. It is estimated that approximately 1.3 billion people are at high risk in endemic cholera countries [2]. The periodically occurring cholera epidemics and their local outbreaks in Africa, South Asia, and Southeast Asia are of great concern. Moreover, the appearance of multidrug-resistant V. cholerae strains replacing antibiotic-susceptible strains [3] increases the risk of high severity of future cholera epidemics and outbreaks. Therefore, V. cholerae represents a global health threat, stimulating the development of public health measures to limit the spread of pathogenic strains to prevent cholera epidemics. Long-term efforts to improve water quality, sanitation, and hygiene are not enough for successful cholera control in developing countries. Vaccination represents a highly effective short- or medium-term strategy for cholera control. Currently, vaccination with oral cholera vaccines (OCVs) is considered by the World Health Organization to be an essential tool in cholera outbreak prevention and control. However, clinical studies have revealed lower protectiveness of OCVs in children <5 years of age compared with older individuals [4]. Moreover, live attenuated V. cholerae strains show better protection and efficacy [5][6][7]. In addition, two- or higher-dose vaccine regimens, although more effective, are more expensive and less feasible than a single-dose regimen. These data indicate the need for further improvement of cholera vaccines.
Genetic engineering represents one of the ways to improve cholera vaccine strains. Genetically engineered vaccine strains can be divided into two groups. The first group comprises attenuated pathogenic strains that lack toxic genes. The second group comprises natural nontoxigenic strains expressing immunogenic but nonactive toxic proteins. The genetically attenuated strain HaitiV is an example of the first group [8]. This strain was obtained by rational design of the pathogenic strain of the V. cholerae serovar El Tor, which caused an outbreak in 2010 in Haiti [9]. VA1.3 is an example of the second group and was constructed from the nontoxigenic strain of V. cholerae El Tor, Inaba, by integrating multiple copies of ctxB into the hly locus [10]. Natural nontoxigenic strains have advantages over attenuated pathogenic strains because they are less toxic, require less genetic modification, and usually have active defence systems that protect them from infection by toxigenic phages such as CTX. However, Vibrio strains can be poorly transformable or nontransformable [11] due to the presence of DNases encoded in the genome or by mobile genetic elements [12][13][14]. V. cholerae strains also have active clustered regularly interspaced short palindromic repeats (CRISPR)/Cas systems [15][16] that can also interfere with bacterial transformation [17][18][19].
Here, we developed a strategy for fast engineering of a new V. cholerae candidate vaccine strain using a chromoprotein-based synthetic reporter operon. The strategy consists of the four steps: (1) characterization of natural bacterial isolates to choose the candidate; (2) optimization of the bacterial cell transformation protocol; (3) development of an amilCP-expressing reporter operon; and (4) genome integration of the reporter operon expressing an immuno-stimulating protein with simultaneous deletion of the recA locus.
2. Identification and Initial Characterization of Natural V. cholerae Isolates
In the first step of our strategy for creating a candidate vaccine strain, we used several methods to identify and characterize two natural nontoxigenic V. cholerae strains, designated strains 31 and 41. According to the MALDI-TOF mass spectrometry results obtained with a MALDI BioTyper, strains 31 and 41 were identified as Vibrio albensis (with identification scores equal to 2.01 and 1.88, respectively). V. albensis is the name for species comprising non-O1 and non-O139 V. cholerae strains [20][21]. However, the identification scores allow us only to reliably conclude that both strains belong to the Vibrio genus. Although microscopic examination and sugar utilization tests also indicated that our strains have Vibrio-like features, these results were not enough to identify the strains. To further increase the identification reliability, we sequenced the genomes of our strains on the Ion S5XL platform (Thermo Scientific, Waltham, MA, USA). 16S RNA sequences extracted from the assembled genomes suggested that both strains belonged to the genus Vibrio. Next, a genome-based identification of strains was performed using the maximum average nucleotide identity (ANI) obtained from paired comparisons with all the non-redundant genomes available in the MIGA and the NCBI Genome databases using the NCBI Prok tool on the Microbial Genomes Atlas web server [22]. The genomes of strains 31 and 41 were most similar to those of V. cholerae LMA3984 (ANI 98.95%) and V. cholerae NZ (ANI 98.77%), respectively. Then, an MLST analysis performed with the MLST 2.0 web server showed that both strains belong to V. cholerae. Strain 31 has the sequence type 760, while strain 41 has a new combination of alleles, and the nearest sequence type is 1317. Thus, genetic analysis strongly suggested that our strains belong to the species V. cholerae.
Next, draft genome sequences were used to search for the presence of CRISPR/Cas systems using CRISPRminer [23]. We found CRISPR/Cas components of type IF systems with spacers against various V. cholerae phages in both strains. Consistently, only fragments of prophages were present in the strains’ genomes. Moreover, as indicated in the strains’ passports, they are resistant to various phages. Therefore, the data suggest that the identified CRISPR/Cas systems are active.
Then, we searched for virulence genes and genes that provide resistance to known antibiotics. In the case of strain 31, 148 factors with varying degrees of integrity of the reading frame were found. In the case of strain 41, 140 factors were found. According to the BLAST search results, the strains have some genes encoding accessory toxins. Most of these genes are inactivated by indels, while some of them seem to be active. Therefore, the ORF of the hlyA gene encoding haemolysin in strain 41 has no errors, which is consistent with the formation of large α-haemolysis zones. In contrast, the hlyA gene in strain 31 was inactivated by indels, and accordingly, the colonies formed weak β-haemolysis zones. We concluded that strain 31 is safer and used it in subsequent experiments.
3. Optimization of V. cholerae Transformation
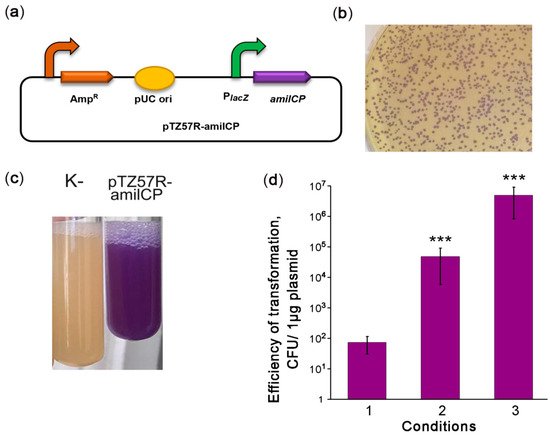
Highly efficient transformation is essential for effective genetic engineering of a chosen bacterial strain. In the second step of our strategy, we optimized the transformation of V. cholerae strain 31. Therefore, we constructed the reporter plasmid pTZ57R-amilCP encoding the amilCP purple chromoprotein (CP) and having an E. coli pUC origin (Figure 1a) that can be maintained in Vibrio species [12][24]. Using the protocol for V. cholerae transformation by electroporation as described in [25], we obtained 101–102 CFU per 1 μg of plasmid (Figure 1b, condition 1). However, this transformation efficiency was not enough to modify the V. cholerae genome with an integrative construct. We hypothesize that the low transformation efficiency may be caused by DNA degradation. In the protocol [25], the cells are used in the logarithmic growth phase, when V. cholerae secretes two extracellular DNases, Dns and Xds. Dns endonuclease is responsible for plasmid degradation [26], while Xds endonuclease is responsible for the degradation of linear DNA [27]. The genome of the V. cholerae 31 strain contains complete sequences for genes encoding both DNases. Dns is repressed in the stationary phase by a quorum sensing-dependent mechanism [26][28]. Therefore, we used a stationary phase culture for transformation. This allowed us to increase the transformation efficiency significantly to approximately 105–106 CFU per 1 μg of the plasmid (Figure 1b, condition 2). Using cells grown to the stationary phase on minimal M9 medium further increased the efficiency of V. cholerae transformation to 106–107 CFU per 1 μg of plasmid (Figure 1b condition 3). Thus, we optimized the conditions for the high-efficiency transformation of natural V. cholerae strains by electroporation.
Figure 1. Optimization of V. cholerae electrotransformation. (a) Scheme of the episomal reporter plasmid pTZ57R-amilCP. PlacZ: promoter of the LacZ α-peptide, amilCP: the gene encoding the purple chromoprotein. The curved arrows denote promoters; (b) colonies of V. cholerae transformed with pTZ57R-amilCP; (c) stationary-phase liquid cultures of V. cholerae strains that were nontransformed (K-) or transformed with the plasmid pTZ57R-amilCP; and (d) improvement of the protocol for V. cholerae transformation by electroporation. CFU: colony-forming unit. The conditions for transformation are described in the text. The data are presented as the mean (n = 3) ± SD. Statistical significance was calculated using Student’s two-tailed t-test for comparing two independent means. *** indicates p < 0.001.
4. Optimization of Synthetic Reporter Operon Expression
In the third step, we optimized the expression of the amilCP-based reporter operon. The promoter dramatically affects heterologous protein expression in V. cholerae both in vitro and in vivo [29][30][31]. Previously, the V. cholerae lacZ locus was used as a safe site for integrating genetic constructs [32][33], and the lacZ promoter was utilized to express heterologous proteins [34]. Therefore, we created the integration construct pCI-amilCP for insertion of amilCP into the lacZ locus (Figure 2a). This construct harboured the amilCP-lacZ synthetic operon under the control of the natural lacZ promoter. Integration flanks were obtained from the other V. cholerae strain, P-19241. Since they differ in sequence from the V. cholerae strain 31 lacZ locus, we identified the part of the lacZ locus that came from the integration plasmid. The integration flanks were 3 kb long; flanks of such length provide high integration efficiency [35]. After pCI-amilCP transformation, none of the grown colonies had colour imparted by amilCP. We hypothesized that the amilCP expression level from the integrated construct was not sufficient to colourize the colonies.
Figure 2. Integration of the synthetic reporter operon into the lacZ locus. (a) Schematic depiction of the integration reporter plasmid pCI-amilCP. The curved arrows denote promoters; (b) schematic depiction of the integrative pALAL plasmid carrying the synthetic reporter operon containing the genes for β-lactamase (AmpR), E. coli galactopermease (lacY), chromoprotein (amilCP), and V. cholerae β-galactosidase (lacZ); (c) colour of cell pellets of V. cholerae transformed with the integrative plasmid pALAL. An untransformed V. cholerae culture (K-) was used as a negative control. V. cholerae transformed with the plasmid pTZ57R-amilCP was used as a positive control. (d) Design of PCR to check the correct integration of the reporter operon. The positions of primers are marked with arrows. If the construct is integrated at the correct genomic position, the primer pair VC-17/VC-18 should yield a 3903 bp fragment, and the VC-19/VC-20 pair should yield a 3182 bp fragment; (e) PCR analysis of the edited V. cholerae colonies. PCR fragments were separated in a 1.5% agarose gel in the presence of EtBr. Genomic DNA from nontransformed strain 31 was used as a negative control (K-). M: 1 kb DNA marker, 1–15: colonies used in the analysis. An asterisk (*) indicates the non-specific band.
Next, we tried to use lacZ as a reporter gene. The genome of strain 31 harbours a complete β-galactosidase gene without deleterious mutations. Accordingly, the ONPG test confirmed the presence of β-galactosidase activity in the cell lysates. As expected for V. cholerae strains, the genome of our strain has no gene for high-affinity 5-Bromo-4-chloro-3-indoyl-beta-D-galactopyranoside (X-Gal) transport, such as LacY galactopermease. Unexpectedly, strain 31 was able to utilize X-Gal. The colonies developed colour slowly within 48 h, suggesting the presence of some low-activity transporters for X-Gal. The possible transporters for X-Gal can be ABC transporters encoded by the mgl operon and involved in methylgalactoside and galactose transport in gram-negative bacteria [36][37]. Previously, the E. coli lacYZ operon was used as a strong reporter for in vivo identification of infection-inducing genes in pathogenic V. cholerae strains [38]. Therefore, we took advantage of active β-galactosidase and added the E. coli lacY gene to the synthetic reporter construct (Figure 2b). Genes were separated by spacers from the V. cholerae S10 operon of the ribosomal proteins. We reasoned that S10 spacers should provide a high level of mRNA translation. We expected that colonies expressing the improved reporter operon would develop colour more rapidly. Indeed, in E. coli, the lacY-containing synthetic reporter causes E. coli colonies to develop more intense colouration. However, the improved reporter operon did not enhance V. cholerae transformant colouration intensity on X-Gal plates (data not shown). We hypothesize that X-Gal transport is not the rate-limiting process of colour development in our strain.
To grow only transformed colonies, we added the β-lactamase gene into the synthetic reporter operon, yielding the pALAL integrative plasmid (Figure 2b). The β-lactamase gene should mediate resistance to ampicillin in transformed cells that are otherwise sensitive to it. V. cholerae transformation with the pALAL plasmid provided 101–102 colonies per 1 μg of the plasmid on ampicillin-supplemented agar plates. The cell pellets of several randomly picked transformed colonies 2, 3, and 5 clearly had a darker colour than control non-transformed cells, suggesting a low level of amilCP accumulation (Figure 2c). PCR with pairs of the primers VC-17/VC-18 and VC-19/VC-20 verified the integration of the reporter operon into the target lacZ locus in these and other recombinant V. cholerae colonies (Figure 2d,e). Sequencing of PCR products further confirmed the correct integration of constructs.
Moreover, we also tried to transform V. cholerae with linear DNA. Therefore, we used a 9082 bp integration cassette amplified from the pALAL plasmid with the primers VC05 and VC09. As a result, we obtained only one ampicillin-resistant colony (# 1). This colony had no purple colour. However, the cell pellet from this colony had a darker colour than that in the negative control (Figure 2c), suggesting a low level of amilCP accumulation. PCR confirmed correct integration of the construct in the lacZ locus (Figure 2e). Unexpectedly, sequencing revealed the integration of a 516 bp fragment of the p15A plasmid ori next to the left flank of the integrated construct. We hypothesize that the PCR fragment was destroyed upon transformation (apparently, by the Xds exonuclease). Seemly, the pALAL plasmid copurified with the PCR product was integrated into the genome instead of the PCR product. Therefore, we may conclude that our strain can be transformed only by circular plasmids.
Our results suggest that the native lacZ promoter is relatively weak and cannot provide sufficient expression levels of reporter proteins to visualize transformed colonies. Therefore, we used the stronger promoter of recA, which highly and stably expresses the housekeeping gene in V. cholerae [31]. We assembled two plasmids bearing a synthetic operon consisting of the amilCP and chloramphenicol acetyltransferase genes under the control of the recA promoter and terminator regions. The plasmid pCI-RACR-0.5 bears short recA flanks that are 0.5 kb long (Figure 3a) and is supposed to be episomal, while the pCI-RACR-3.0 plasmid bears 3 kb-long flanks (Figure 3b) and is expected to be integrated into the recA locus. As expected, pCI-RACR-0.5 was not integrated into the recA locus. This was confirmed by PCR (Figure 3c,d) and the presence of the episomal plasmid in cell cultures (Figure 3e). The V. cholerae colonies, as in the case of pTZ57R-amilCP, were coloured purple. V. cholerae colonies transformed with pCI-RACR-3.0 exhibited a purple centre (Figure 3f), and their pellets were also clearly purple in colour (Figure 3g). PCR confirmed the correct integration of pCI-RACR-3.0 into the recA locus (Figure 3h). Next, we found that V. cholerae colonies grown on brain heart infusion (BHI) medium for 72 h developed a more intense colour. Therefore, we optimized the expression of the synthetic operon to identify transformed V. cholerae colonies simply by visual inspection.
Figure 3. Integration of the synthetic reporter operon into the recA locus. (a,b) Schematic depiction of the pCI-RACR reporter constructs. The curved arrows denote promoters; (c) scheme of the experiment for PCR verification of construct integration. The positions of primers are marked with arrows; (d) PCR check for the integration of pCI-RACR-0.5. If the construct was integrated into the correct genomic position, the VC-43/VC-44 pair amplified a fragment of 3157 bp (lane 1), the VC-43/VC-47 pair amplified a fragment of 3330 bp (lane 2), the VC-43/VC-34 pair amplified a fragment of 3824 bp (lane 3), the VC-45/VC-46 pair amplified a fragment of 3250 bp (lane 4), and the VC-35/VC-46 pair amplified a fragment of 3929 bp (lane 5). (e) Plasmid preparations from E. coli (lane 1) or V. cholerae colonies transformed with pCI-RACR-0.5 (lanes 2 and 3); (f) colour of V. cholerae colonies transformed with the pALAL or pCI-RACR-3.0 integrative plasmid; (g) colour of cell pellets of V. cholerae transformed with the integrative pCI-CR-3.0. An untransformed V. cholerae culture (K-) was used as a negative control; (h) PCR check for the integration of pCI-RACR-3.0. Untransformed V. cholerae strain 31 (K-) was used as a negative control. The primers used were the same as those described in part (c) of the figure.
5. Assessing the Stability of the Genetic Constructs
Colony colouration due to the expression of amilCP as a reporter protein is the fastest and easiest way to monitor the presence of genetic constructs and track their stability. To this end, we used episomal pCI-RACR-0.5 and integrative pCI-RACR-3.0 plasmids. The scheme for the stability test is presented in Figure 4a. According to the stability test results (Figure 4b), the colonies with the integrated construct displayed only a subtle decrease in the percentage of coloured and Cm-resistant colonies: 93.6 and 95.2% of the corresponding colonies were observed after the 3rd passage. Simultaneously, the colonies transformed with the episomal plasmid showed a dramatic decrease in coloured and Cm-resistant colonies: 1.3% of coloured colonies were observed after the 3rd passage. Generally, the number of antibiotic-resistant colonies was higher than the number of coloured colonies. This was more obvious for colonies transformed with the episomal plasmid. We hypothesize that the copy number of genes producing an enzyme can be as low as one or two to provide cellular resistance to antibiotics. In contrast, the copy number should be significantly high to provide enough CP expression to colourize the cells. We observed the same situation for the integrative construct when the copy number was not enough to colourize the colonies but enough to provide ampicillin resistance. We then observed the sectoral colouring (Figure 4c). The colourless sectors seemingly appeared due to plasmid loss or a rapid decrease in the plasmid copy number. These data suggest that the integrated construct was highly stable, while the plasmid was rapidly lost. Notably, in the presence of Cm, only purple colonies grew, suggesting that both genetic constructs were stable under antibiotic selection.
Figure 4. A test for stability of the genetic constructs. (a) Scheme of the experiment for assessing the stability of the genetic constructs. The chosen colonies were grown in BHI medium without antibiotics for 16 h at RT. Equal volumes of cultures of the same passages were diluted 106- or 107-fold, plated on BHI plates with or without Cm, and incubated at RT for 72 h. One-third of the first overnight culture was transferred to a tube with fresh BHI medium without antibiotics and grown for 24 h at RT. Then, the plating of the grown cultures was repeated. (b) Quantitative data on the genetic construct stability test. V. cholerae cultures grown to the stationary phase were diluted 106-fold (1st and 3rd passages) or 107-fold (2nd passage), plated, and grown on BHI agar plates. Colonies were counted using a Clono Counter [39] or manually depending on the number of colonies to be counted. The number of coloured colonies grown in the presence of Cm was set to 100%. The data are presented as the mean (n = 5) ± SD. Statistical significance: NS: nonsignificant differences, * indicates p between 0.05 and 0.01, *** indicates p < 0.001 according to one-way ANOVA test; (c) colour heterogeneity of V. cholerae colonies transformed with the plasmid. After the first passage, V. cholerae cultures were diluted 106-fold, spread on BHI agar plates without antibiotics, and incubated for 72 h at RT.
6. Construction of a Candidate V. cholerae Vaccine Strain
In the final step, we proceeded to construct a candidate vaccine strain. Since the cholera toxin β-subunit (CtxB) is safe and provides protective properties for vaccines, e.g., for the widely used rBS-WC (Dukoral) [40][41][42], we sought to convert our strain to a CtxB producer. To achieve this goal, we assembled constructs to integrate a synthetic operon consisting of ctxB, amilCP, and cat into the recA locus. However, in all the constructs purified from violet E. coli colonies resistant to Cm, the ctxB ORF was damaged, leading to inactive protein synthesis. We hypothesize that high levels of the cholera toxin produced from the strong constitutive promoter are highly toxic to E. coli. Therefore, the surviving colonies harboured the assembled constructs with ctxB mutations. We reasoned that to assemble the construct correctly, we needed to use a promoter that is weak or inactive in E. coli and active in V. cholerae. The promoter of the cholera toxin operon is very weak in E. coli [43]. At the same time, it is highly active in V. cholerae and directly regulated by two transcription factors, toxT [44] and toxR [45]. Indeed, the construct (Figure 5a) was assembled in E. coli correctly, suggesting that the β-subunit was expressed at nontoxic levels. The obtained integration construct was transformed into V. cholerae. The colony with the most intense violet colouration was further analysed. PCR confirmed the correct integration of the ctxB-containing operon into the recA locus (Figure 5b). The recombinant strain produced the cholera toxin β-subunit at 6.64 µg/mL in the cell lysate and about 1.6 µg/mL in the culture media (Figure 5c). The productivity of our strain is close to one of the best CTB-producing V. cholerae recombinant strains M7922-C1, which produces β-subunit at 3.17 ± 1.69 µg/mL [46]. The genetic stability test suggests the high stability of the integrated ctxB-expressing operon (Figure 5d). Thus, we constructed a novel V. cholerae candidate vaccine strain designated rVCH-31.1 suitable for further preclinical studies.
Figure 5. Construction of a candidate V. cholerae vaccine strain expressing cholera toxin β-subunit. (a) Schematic depiction of the pCI-RCCACR-3.0 integrative construct bearing a synthetic operon with ctxB, the amilCP reporter, and the cat marker genes. The curved arrows denote promoters; (b) PCR check for the integration of pCI-RCCACR-3.0. The positions of the primers are marked with arrows. In the case of correct genomic integration of the construct, the VC-43/VC-44 primer pair amplified a fragment of 3157 bp (lane 1), the VC-43/VC-39 primer pair amplified a fragment of 3368 bp (lane 2), the VC-43/VC-41 primer pair amplified a fragment of 3759 bp (lane 3), the VC-45/VC-46 primer pair amplified a fragment of 3250 bp (lane 4), and the VC-35/VC-46 primer pair amplified a fragment of 3929 bp (lane 5). M: DNA molecular weight marker; (c) results of GM1-ELISA of CtxB production by the rVCH-31.1 strain. The data are presented as the mean (n = 3) ± SD. The negative controls were the original V. cholerae nontoxigenic strain (K-1) or recombinant strain edited with the pCI-RACR-3.0 plasmid (K-2). ‘<Curve’ indicates that the signal was below the calibration curve; (d) quality control of the V. cholerae candidate vaccine strain by the genetic stability test. The data are presented as the mean (n = 5) ± SD. NS: nonsignificant differences according to one-way ANOVA.
References
- Clemens, J.D.; Nair, G.B.; Ahmed, T.; Qadri, F.; Holmgren, J. Cholera. Lancet 2017, 390, 1539–1549.
- Shaikh, H.; Lynch, J.; Kim, J.; Excler, J.L. Current and future cholera vaccines. Vaccine 2020, 38, A118–A126.
- Weill, F.X.; Domman, D.; Njamkepo, E.; Tarr, C.; Rauzier, J.; Fawal, N.; Keddy, K.H.; Salje, H.; Moore, S.; Mukhopadhyay, A.K.; et al. Genomic history of the seventh pandemic of cholera in Africa. Science 2017, 358, 785–789.
- World Health Organization. Cholera vaccines: WHO position paper. In Weekly Epidemiological Record = Relevé Épidémiologique Hebdomadaire; WHO: Geneva, Switzerland, 2010; Volume 85, pp. 117–128.
- Chen, W.H.; Cohen, M.B.; Kirkpatrick, B.D.; Brady, R.C.; Galloway, D.; Gurwith, M.; Hall, R.H.; Kessler, R.A.; Lock, M.; Haney, D.; et al. Single-dose Live Oral Cholera Vaccine CVD 103-HgR protects against human experimental infection with vibrio cholerae O1 El Tor. Clin. Infect. Dis. 2016, 62, 1329–1335.
- Qadri, F.; Chowdhury, M.I.; Faruque, S.M.; Salam, M.A.; Ahmed, T.; Begum, Y.A.; Saha, A.; Al Tarique, A.; Seidlein, L.V.; Park, E.; et al. Peru-15, a live attenuated oral cholera vaccine, is safe and immunogenic in Bangladeshi toddlers and infants. Vaccine 2007, 25, 231–238.
- Calain, P.; Chaine, J.P.; Johnson, E.; Hawley, M.L.; O’Leary, M.J.; Oshitani, H.; Chaignat, C.L. Can oral cholera vaccination play a role in controlling a cholera outbreak? Vaccine 2004, 22, 2444–2451.
- Hubbard, T.P.; Billings, G.; Dorr, T.; Sit, B.; Warr, A.R.; Kuehl, C.J.; Kim, M.; Delgado, F.; Mekalanos, J.J.; Lewnard, J.A.; et al. A live vaccine rapidly protects against cholera in an infant rabbit model. Sci. Transl. Med. 2018, 10, eaap8423.
- Chin, C.S.; Sorenson, J.; Harris, J.B.; Robins, W.P.; Charles, R.C.; Jean-Charles, R.R.; Bullard, J.; Webster, D.R.; Kasarskis, A.; Peluso, P.; et al. The origin of the Haitian cholera outbreak strain. N. Engl. J. Med. 2011, 364, 33–42.
- Thungapathra, M.; Sharma, C.; Gupta, N.; Ghosh, R.K.; Mukhopadhyay, A.; Koley, H.; Nair, G.B.; Ghosh, A. Construction of a recombinant live oral vaccine from a non-toxigenic strain of Vibrio cholerae O1 serotype inaba biotype E1 Tor and assessment of its reactogenicity and immunogenicity in the rabbit model. Immunol. Lett. 1999, 68, 219–227.
- Simpson, C.A.; Podicheti, R.; Rusch, D.B.; Dalia, A.B.; van Kessel, J.C. Diversity in natural transformation frequencies and regulation across vibrio species. mBio 2019, 10, e02788-19.
- Panda, D.K.; Dasgupta, U.; Das, J. Transformation of Vibrio cholerae by plasmid DNA. Gene 1991, 105, 107–111.
- Marcus, H.; Ketley, J.M.; Kaper, J.B.; Holmes, R.K. Effects of DNase production, plasmid size, and restriction barriers on transformation of Vibrio cholerae by electroporation and osmotic shock. FEMS Microbiol. Lett. 1990, 56, 149–154.
- Dalia, A.B.; Seed, K.D.; Calderwood, S.B.; Camilli, A. A globally distributed mobile genetic element inhibits natural transformation of Vibrio cholerae. Proc. Natl. Acad. Sci. USA 2015, 112, 10485–10490.
- McDonald, N.D.; Regmi, A.; Morreale, D.P.; Borowski, J.D.; Boyd, E.F. CRISPR-Cas systems are present predominantly on mobile genetic elements in Vibrio species. BMC Genom. 2019, 20, 105.
- Bourgeois, J.; Lazinski, D.W.; Camilli, A. Identification of spacer and protospacer sequence requirements in the vibrio cholerae type I-E CRISPR/Cas system. mSphere 2020, 5, e00813–e00820.
- Bikard, D.; Hatoum-Aslan, A.; Mucida, D.; Marraffini, L.A. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 2012, 12, 177–186.
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71.
- Zhang, Y.; Heidrich, N.; Ampattu, B.J.; Gunderson, C.W.; Seifert, H.S.; Schoen, C.; Vogel, J.; Sontheimer, E.J. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol. Cell 2013, 50, 488–503.
- Hasan, N.A.; Rezayat, T.; Blatz, P.J.; Choi, S.Y.; Griffitt, K.J.; Rashed, S.M.; Huq, A.; Conger, N.G.; Colwell, R.R.; Grimes, D.J. Nontoxigenic Vibrio cholerae non-O1/O139 isolate from a case of human gastroenteritis in the U.S. Gulf Coast. J. Clin. Microbiol. 2015, 53, 9–14.
- Kasai, S. Freshwater bioluminescence in Vibrio albensis (Vibrio cholerae biovar albensis) NCIMB 41 is caused by a two-nucleotide deletion in luxO. J. Biochem. 2006, 139, 471–482.
- Rodriguez, R.L.; Gunturu, S.; Harvey, W.T.; Rossello-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The microbial genomes atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288.
- Zhang, F.; Zhao, S.; Ren, C.; Zhu, Y.; Zhou, H.; Lai, Y.; Zhou, F.; Jia, Y.; Zheng, K.; Huang, Z. CRISPRminer is a knowledge base for exploring CRISPR-Cas systems in microbe and phage interactions. Commun. Biol. 2018, 1, 180.
- Weinstock, M.T.; Hesek, E.D.; Wilson, C.M.; Gibson, D.G. Vibrio natriegens as a fast-growing host for molecular biology. Nat. Methods 2016, 13, 849–851.
- Gonzales, M.F.; Brooks, T.; Pukatzki, S.U.; Provenzano, D. Rapid protocol for preparation of electrocompetent Escherichia coli and Vibrio cholerae. J. Vis. Exp. 2013, e50684.
- Seper, A.; Fengler, V.H.; Roier, S.; Wolinski, H.; Kohlwein, S.D.; Bishop, A.L.; Camilli, A.; Reidl, J.; Schild, S. Extracellular nucleases and extracellular DNA play important roles in Vibrio cholerae biofilm formation. Mol. Microbiol. 2011, 82, 1015–1037.
- Pressler, K.; Mitterer, F.; Vorkapic, D.; Reidl, J.; Oberer, M.; Schild, S. Characterization of Vibrio cholerae’s extracellular nuclease Xds. Front. Microbiol. 2019, 10, 2057.
- Blokesch, M.; Schoolnik, G.K. The extracellular nuclease Dns and its role in natural transformation of Vibrio cholerae. J. Bacteriol. 2008, 190, 7232–7240.
- John, M.; Crean, T.I.; Calderwood, S.B.; Ryan, E.T. In vitro and in vivo analyses of constitutive and in vivo-induced promoters in attenuated vaccine and vector strains of Vibrio cholerae. Infect. Immun. 2000, 68, 1171–1175.
- Morin, C.E.; Kaper, J.B. Use of stabilized luciferase-expressing plasmids to examine in vivo-induced promoters in the Vibrio cholerae vaccine strain CVD 103-HgR. FEMS Immunol. Med. Microbiol. 2009, 57, 69–79.
- Lo Scrudato, M.; Blokesch, M. The regulatory network of natural competence and transformation of Vibrio cholerae. PLoS Genet. 2012, 8, e1002778.
- Taylor, D.N.; Killeen, K.P.; Hack, D.C.; Kenner, J.R.; Coster, T.S.; Beattie, D.T.; Ezzell, J.; Hyman, T.; Trofa, A.; Sjogren, M.H.; et al. Development of a live, oral, attenuated vaccine against El Tor cholera. J. Infect. Dis. 1994, 170, 1518–1523.
- Ryan, E.T.; Butterton, J.R.; Zhang, T.; Baker, M.A.; Stanley, S.L.; Calderwood, S.B. Oral immunization with attenuated vaccine strains of Vibrio cholerae expressing a dodecapeptide repeat of the serine-rich Entamoeba histolytica protein fused to the cholera toxin B subunit induces systemic and mucosal antiamebic and anti-V. cholerae antibody responses in mice. Infect. Immun. 1997, 65, 3118–3125.
- Butterton, J.R.; Beattie, D.T.; Gardel, C.L.; Carroll, P.A.; Hyman, T.; Killeen, K.P.; Mekalanos, J.J.; Calderwood, S.B. Heterologous antigen expression in Vibrio cholerae vector strains. Infect. Immun. 1995, 63, 2689–2696.
- Marvig, R.L.; Blokesch, M. Natural transformation of Vibrio cholerae as a tool—Optimizing the procedure. BMC Microbiol. 2010, 10, 155.
- Harayama, S.; Bollinger, J.; Iino, T.; Hazelbauer, G.L. Characterization of the mgl operon of Escherichia coli by transposon mutagenesis and molecular cloning. J. Bacteriol. 1983, 153, 408–415.
- Muller, N.; Heine, H.G.; Boos, W. Characterization of the Salmonella typhimurium mgl operon and its gene products. J. Bacteriol. 1985, 163, 37–45.
- Camilli, A.; Mekalanos, J.J. Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol. Microbiol. 1995, 18, 671–683.
- Niyazi, M.; Niyazi, I.; Belka, C. Counting colonies of clonogenic assays by using densitometric software. Radiat Oncol 2007, 2, 4.
- Clemens, J.D.; Sack, D.A.; Harris, J.R.; Van Loon, F.; Chakraborty, J.; Ahmed, F.; Rao, M.R.; Khan, M.R.; Yunus, M.; Huda, N.; et al. Field trial of oral cholera vaccines in Bangladesh: Results from three-year follow-up. Lancet 1990, 335, 270–273.
- Lucas, M.E.; Deen, J.L.; von Seidlein, L.; Wang, X.Y.; Ampuero, J.; Puri, M.; Ali, M.; Ansaruzzaman, M.; Amos, J.; Macuamule, A.; et al. Effectiveness of mass oral cholera vaccination in Beira, Mozambique. N. Engl. J. Med. 2005, 352, 757–767.
- Jelinek, T.; Kollaritsch, H. Vaccination with Dukoral against travelers’ diarrhea (ETEC) and cholera. Expert Rev. Vaccines 2008, 7, 561–567.
- Miller, V.L.; Mekalanos, J.J. Synthesis of cholera toxin is positively regulated at the transcriptional level by toxR. Proc. Natl. Acad. Sci. USA 1984, 81, 3471–3475.
- DiRita, V.J.; Parsot, C.; Jander, G.; Mekalanos, J.J. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 1991, 88, 5403–5407.
- Hung, D.T.; Mekalanos, J.J. Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc. Natl. Acad. Sci. USA 2005, 102, 3028–3033.
- Rhie, G.E.; Jung, H.M.; Park, J.; Kim, B.S.; Mekalanos, J.J. Construction of cholera toxin B subunit-producing Vibrio cholerae strains using the Mariner-FRT transposon delivery system. FEMS Immunol. Med. Microbiol. 2008, 52, 23–28.
More
Information
Subjects:
Health Care Sciences & Services
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
651
Revisions:
2 times
(View History)
Update Date:
22 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No