 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kirsty J McLean | + 5148 word(s) | 5148 | 2021-10-25 10:45:40 | | | |
| 2 | Vivi Li | Meta information modification | 5148 | 2021-11-03 02:58:22 | | |
Video Upload Options
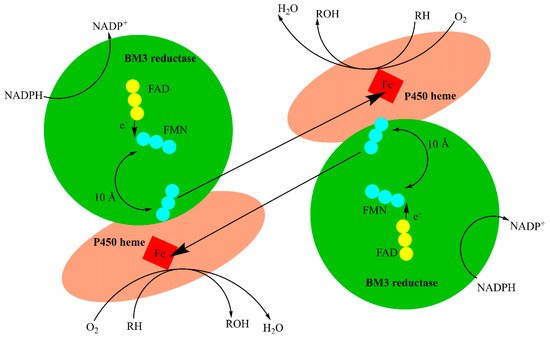
CYP102A1 (BM3) is a catalytically self-sufficient flavocytochrome fusion protein isolated from Bacillus megaterium, which displays similar metabolic capabilities to many drug-metabolizing human P450 isoforms. BM3′s high catalytic efficiency, ease of production and malleable active site makes the enzyme a desirable tool in the production of small molecule metabolites, especially for compounds that exhibit drug-like chemical properties. The engineering of select key residues within the BM3 active site vastly expands the catalytic repertoire, generating variants which can perform a range of modifications. This provides an attractive alternative route to the production of valuable compounds that are often laborious to synthesize via traditional organic means.
1. Context and Introduction to BM3
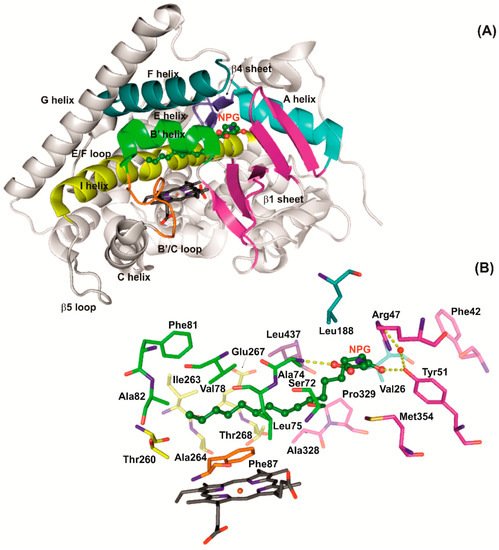
P450 BM3
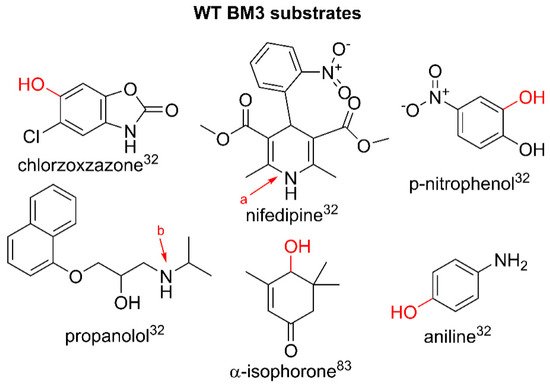
2. Wild Type BM3 and Pharmaceutical Metabolism
3. Engineering BM3 for Drug Metabolite Production
3.1. Key Promiscuous BM3 Variants and Library Screening
| BM3 Variant | Point Mutations | Substrates |
|---|---|---|
| LVQ [39] | R47L/F87V/L188Q | clozapine [39], diclofenac [39], acetaminophen [39], omeprazole [43], reservatrol * [44], polydatin [45][46], phloretin * [47][48], cladosporin [49] |
| M01 [39] | LVQ/E267V/G415S/G1049E | clozapine [39], diclofenac [39], acetaminophen [39], testosterone * [50], noresthisterone * [51], methoxyresorufin [52], ethoxyresorufin [52] |
| M02 [39] | LVQ/L86I/N319T/A964V | clozapine [39], diclofenac [39], acetaminophen [39], norandrostenedione [53], methoxyresorufin [52], ethoxyresorufin [52], buspirone [52], amitriptyline [52], aripiprazole [52] |
| M05 [39] | LVQ/F81I/E267V/G415S | clozapine [39], diclofenac [39], acetaminophen [39], methoxyresorufin [52], ethoxyresorufin [52] |
| M11 [39] | LVQ/E64G/F81I/E143G/E267V/G415S | clozapine [39], diclofenac [39], acetaminophen [39], Testosterone * [50][54], norethisterone * [51][54], dextromethorphan [40], nifedipine [40], midazolam [40], 3,4-methylenedioxymethylamphetamine [40], estradiol [14][55], methoxyresorufin [52], ethoxyresorufin [52], buspirone [52], duloxetine [52], thioridazine [52], ondansetron [52], imatinib [52], omeprazole [52], rosiglitazone [52], paroxetine [52], norethisterone [52], resveratrol [52], dihydrobenzophenone [52], 17α-ethinylestradiol [52], bisphenol A [52], diethylstilbestrol [52], hexestrol [52], estriol [52], benzophenone-3 [52], aldrin [52], testosterone [52], tramadol [52], imidazole [52], ketoconazole [52] |
| 9-10A [36] | R47C/V78A/K94I/P142S/T175I/A184V/F205C /S226R/H236Q/E252G/R255S/A290V/L353V |
verapamil [56], astemizole [56], LY294002 [56] |
| 139-3 [38] | V78A/H138Y/T175I/V178I/A184V/ H236Q/ E252G/R255S/A290V/A295T/L353V |
androstenedione [57] |
| GVQ [41] | A74G/F87V/L188Q | testosterone [42], amodiaquine [42], dextromethorphan [42], acetaminophen [42], 3,4-methylenedioxymethylamphetamine [42] |
3.2. Production of Human P450 Metabolites
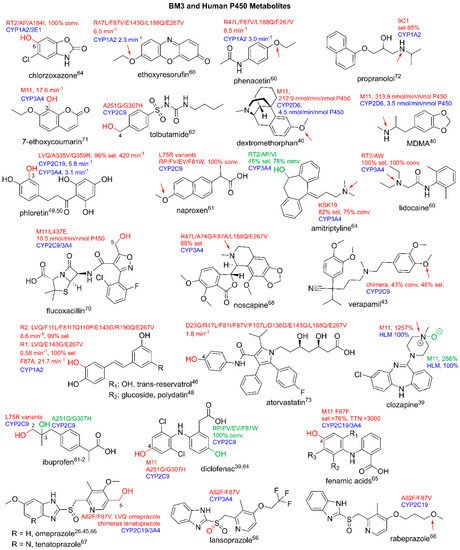
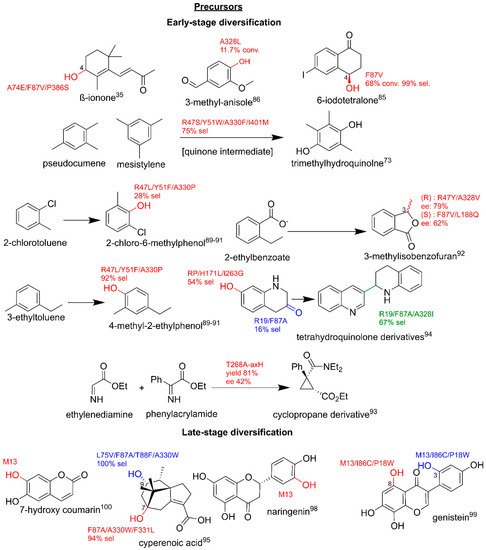
3.3. Production of Clinically Relevant Pharmaceuticals and Related Precursors
3.3.1. Pharmaceutical Intermediate and Final Product Production
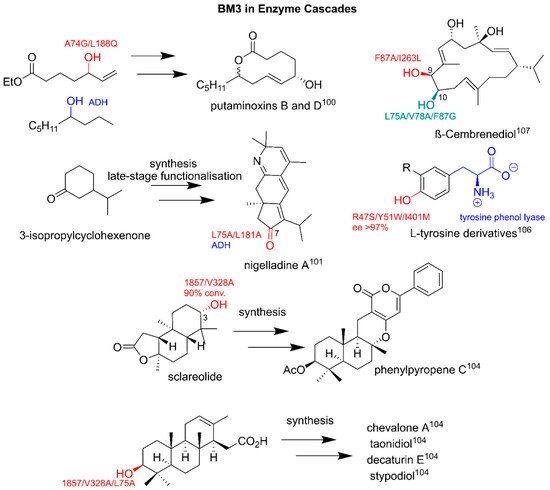
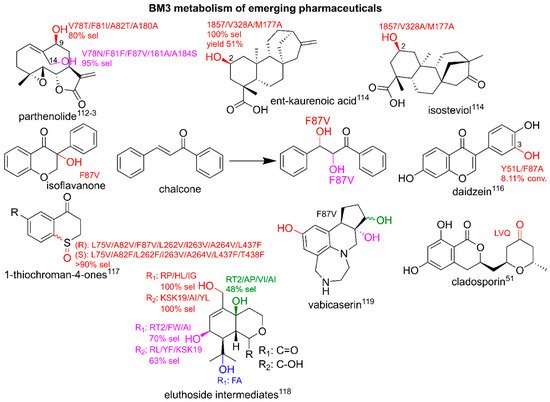
3.4. Utilizing BM3 for Production of Emerging Pharmaceuticals
| Steroid | Structure | OH- Position |
Variant | Parameters |
|---|---|---|---|---|
| Testosterone | 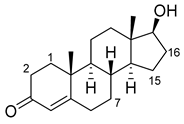 |
2β * | RLYF/KSK19 * [64] | 100% conv, 61% sel |
| 1β | H171L/Q307H/N319Y/F87A/T260G/P329G/A330W [94] | 71% sel, 76% conv | ||
| 7β | R47W/S72W/F77Y/V78L/F81I/A82L/T88S/M177T/M185Q/L188Q/I209T [95] | 90% sel | ||
| 15β * | KSK19/FV/QP [64] | 96% sel, 83% conv | ||
| 16α * | M11/V87I/S72I [50] M01/A82W/S72I [50] |
90% ee 95% ee |
||
| 16β * | M01/A82W [50] M11/V87I [50] |
100% ee 100% ee |
||
| Estradiol | 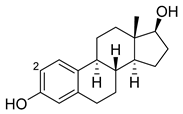 |
2 | M11 [55] | 47 min−1 |
| Norandrostenedione | 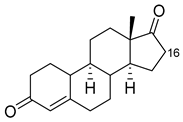 |
16β * | M02 [53] | 95% sel |
| Nandrolone | 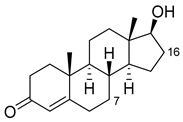 |
7β | R47W/S72W/F77Y/V78L/F81I/A82L/T88S/M177T/M185Q/L188Q/I209T [95] | 75% sel |
| 16α | R47L/S72I/A82F/F87I/L188C/A330W [89] | 98% sel | ||
| 16β | R47W/A82W/F87V/L181Q [89] | 90% sel | ||
| Norethindrone | 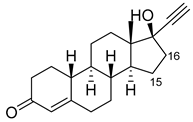 |
15β * | M11/A82W/V87A [51] | 100% sel |
| 16β * | M01/A82W [51] | 96% sel | ||
| Boldenone | 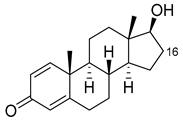 |
16α | R47L/Y51W/S72I/A82W/F87I/L181C [89] | 97% sel |
| 16β | R47W/A82W/F87V/L181Q [89] | 72% sel | ||
| Androstenedione | 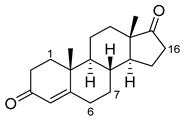 |
1α | 139-3 [57] | |
| 6β | H171L/Q307H/N319Y/F87V/I263G [94] | 78% sel, 29% conv | ||
| 7β | R47W/S72W/F77Y/V78L/F81I/A82L/T88S/M177T/M185Q/L188Q/I209T [95] R47L/Y51F/H171L/Q307H/N319Y/ F87A/A184I/T260G/A328G [94] |
90% sel 81% sel, 94% conv |
||
| 16α | R47W/Y51W/S72I/A82F/F87I/L181C [89] | 95% sel | ||
| 16β | R47W/Y51H/A82W/F87V/L181Q [89] | 100% sel |
References
- Roberts, A.A.; Ryan, K.S.; Moore, B.S.; Gulder, T.A.M. Total (Bio)synthesis: Strategies of nature and of chemists. Top. Curr. Chem. 2010, 297, 149.
- Rupasinghe, S.; Schuler, M.A.; Kagawa, N.; Yuan, H.; Lei, L.; Zhao, B.; Kelly, S.L.; Waterman, M.R.; Lamb, D.C. The cytochrome P450 gene family CYP157 does not contain EXXR in the K-helix reducing the absolute Conserved P450 residues to a single cysteine. FEBS Lett. 2006, 580, 6338–6342.
- Guengerich, F.P.; Macdonald, T.L. Chemical mechanisms of catalysis by cytochromes P-450: A unified view. Acc. Chem. Res. 1984, 17, 9–16.
- Whitehouse, C.J.C.; Bell, S.G.; Wong, L.-L. P450 BM3 (CYP102A1): Connecting the dots. Chem. Soc. Rev. 2012, 41, 1218–1260.
- Iyanagi, T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: Implications for detoxification. Int. Rev. Cytol. 2007, 260, 35–112.
- Miura, Y.; Fulco, A.J. (ω-2) Hydroxylation of fatty acids by a soluble system from Bacillus megaterium. J. Biol. Chem. 1974, 249, 1880–1888.
- Miura, Y.; Fulco, A.J. ω-1, ω-2 and ω-3 hydroxylation of long-chain fatty acids, amides and alcohols by a soluble enzyme system from Bacillus megaterium. Biochim. Biophys. Acta 1975, 388, 305–317.
- Hare, R.S.; Fulco, A.J. Carbon monoxide and hydroxymercuribenzoate sensitivity of a fatty acid (ω-2) hydroxylase from Bacillus megaterium. Biochem. Biophys. Res. Commun. 1975, 65, 665–672.
- Narhi, L.O.; Fulco, A.J. Identification and Characterization of two functional domains in cytochrome P-450BM-3, a catalytically self-sufficient monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 1987, 262, 6683–6690.
- Narhi, L.O.; Fulco, A.J. Characterization of a catalytically self-sufficient 119,000-dalton Cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 1986, 261, 7160–7169.
- Porter, T.D. An unusual yet strongly conserved flavoprotein reductase in bacteria and mammals. Trends Biochem. Sci. 1991, 16, 154–158.
- Lewis, D.F.V.; Watson, E.; Lake, B.G. Evolution of the cytochrome P450 superfamily: Sequence alignments and pharmacogenetics. Mutat. Res. 1998, 410, 245–270.
- Fulco, A.J. Chain elongation, 20hydroxylation, and decarboxylation of long chain fatty acids by yeast. J. Biol. Chem. 1967, 242, 3608–3613.
- English, N.; Hughes, V.; Wolf, C.R. Common pathways of cytochrome P450 gene regulation by peroxisome proliferators and barbiturates in Bacillus megaterium ATCC14581. J. Biol. Chem. 1994, 269, 26836–26841.
- Ravichandran, K.G.; Boddupalli, S.S.; Hasemann, C.A.; Peterson, J.A.; Deisenhofer, J. Crystal structure of hemoprotein domain of P450BM-3, a prototype for microsomal P450′s. Science 1993, 261, 731–736.
- Hasemann, C.A.; Kurumbail, R.G.; Boddupalli, S.S.; Peterson, J.A.; Deisenhofer, J. Structure and function of cytochromes P450: A comparative analysis of three crystal structures. Structure 1995, 3, 41–62.
- Anzenbacher, P.; Hudeček, J. Differences in flexibility of active sites of cytochromes P450 probed by resonance raman and UV-Vis absorption spectroscopy. J. Inorg. Biochem. 2001, 87, 209–213.
- Li, H.; Poulos, T.L. Modeling protein–substrate interactions in the heme domain of cytochrome P450BM−3. Acta Crystallogr. Sect. D Biol. Crystallogr. 1995, 51, 21–32.
- Joyce, M.G.; Girvan, H.M.; Munro, A.W.; Leys, D. A single mutation in cytochrome P450 BM3 induces the conformational rearrangement seen upon substrate binding in the wild-type Enzyme. J. Biol. Chem. 2004, 279, 23287–23293.
- Daff, S.N.; Chapman, S.K.; Turner, K.L.; Holt, R.A.; Govindaraj, S.; Poulos, T.L.; Munro, A.W. Redox control of the catalytic cycle of flavocytochrome P-450 BM3. Biochemistry 1997, 36, 13816–13823.
- Gonvindaraj, S.; Li, H.; Poulos, T.L. Flavin supported fatty acid oxidation by the heme domain of Bacillus megaterium cytochrome P450BM-3. Biochem. Biophys. Res. Commun. 1994, 203, 1745–1749.
- Sevrioukova, I.F.; Peterson, J.A. Reaction of carbon-monoxide and molecular-Oxygen with P450terp (CYP108) and P450BM-3 (CYP102). Arch. Biochem. Biophys. 1995, 317, 397–404.
- Neeli, R.; Girvan, H.M.; Lawrence, A.; Warren, M.J.; Leys, D.; Scrutton, N.S.; Munro, A.W. The dimeric form of flavocytochrome P450 BM3 is catalytically functional as a fatty acid hydroxylase. FEBS Lett. 2005, 579, 5582–5588.
- Li, H.; Poulos, T.L. The structure of the cytochrome P450BM-3 haem domain complexed with the fatty acid substrate, palmitoleic acid. Nat. Struct. Biol. 1997, 4, 140–146.
- Noble, M.A.; Miles, C.S.; Chapman, S.K.; Lysek, D.A.; Mackay, A.C.; Reid, G.A.; Hanzlik, R.P.; Munro, A.W. Roles of key active-site residues in flavocytochrome P450 BM3. Biochem. J. 1999, 339, 371–379.
- Butler, C.F.; Peet, C.; Mason, A.E.; Voice, M.W.; Leys, D.; Munro, A.W. Key mutations alter the cytochrome P450 BM3 conformational landscape and remove inherent substrate bias. J. Biol. Chem. 2013, 288, 25387–25399.
- Volz, T.J.; Rock, D.A.; Jones, J.P. Evidence for two different active oxygen species in cytochrome P450 BM3 mediated sulfoxidation and N-Dealkylation Reactions. J. Am. Chem. Soc. 2002, 124, 9724–9725.
- Cryle, M.J.; De Voss, J.J. Is the ferric hydroperoxy species responsible for sulfur oxidation in cytochrome P450s? Angew. Chemie Int. Ed. 2006, 45, 8221–8223.
- Haines, D.C.; Tomchick, D.R.; Machius, M.; Peterson, J.A. Pivotal role of water in the mechanism of P450BM-3. Biochemistry 2001, 40, 13456–13465.
- Ost, T.W.B.; Clark, J.; Mowat, C.G.; Miles, C.S.; Walkinshaw, M.D.; Reid, G.A.; Chapman, S.K.; Daff, S. Oxygen activation and electron transfer in flavocytochrome P450 BM3. J. Am. Chem. Soc. 2003, 125, 15010–15020.
- Ost, T.W.B.; Miles, C.S.; Munro, A.W.; Murdoch, J.; Reid, G.A.; Chapman, S.K. Phenylalanine 393 exerts thermodynamic control over the heme of flavocytochrome P450 BM3. Biochemistry 2001, 40, 13421–13429.
- Di Nardo, G.; Fantuzzi, A.; Sideri, A.; Panicco, P.; Sassone, C.; Giunta, C.; Gilardi, G. Wild-type CYP102A1 as a biocatalyst: Turnover of drugs usually metabolised by human liver enzymes. J. Biol. Inorg. Chem. 2007, 12, 313–323.
- Whitehouse, C.J.C.; Yang, W.; Yorke, J.A.; Tufton, H.G.; Ogilvie, L.C.I.; Bell, S.G.; Zhou, W.; Bartlam, M.; Rao, Z.; Wong, L.L. Structure, electronic properties and catalytic behaviour of an activity-Enhancing CYP102A1 (P450 BM3) Variant. Dalt. Trans. 2011, 40, 10383–10396.
- Li, Q.S.; Schwaneberg, U.; Fischer, M.; Schmitt, J.; Pleiss, J.; Lutz-Wahl, S.; Schmid, R.D. Rational evolution of a medium chain-specific cytochrome P-450 BM-3 variant. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2001, 1545, 114–121.
- Urlacher, V.B.; Makhsumkhanov, A.; Schmid, R.D. Biotransformation of β-ionone by engineered cytochrome P450 BM-3. Appl. Microbiol. Biotechnol. 2006, 70, 53–59.
- Peters, M.W.; Meinhold, P.; Glieder, A.; Arnold, F.H. Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J. Am. Chem. Soc. 2003, 125, 13442–13450.
- Lewis, J.C.; Mantovani, S.M.; Fu, Y.; Snow, C.D.; Komor, R.S.; Wong, C.-H.; Arnold, F.H. Combinatorial alanine substitution enables rapid optimization of cytochrome P450BM3 for selective hydroxylation of large substrates. ChemBioChem 2010, 11, 2502–2505.
- Glieder, A.; Farinas, E.T.; Arnold, F.H. Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 2002, 20, 1135–1139.
- Damsten, M.C.; van Vugt-Lussenburg, B.M.A.; Zeldenthuis, T.; de Vlieger, J.S.B.; Commandeur, J.N.M.; Vermeulen, N.P.E. Application of drug metabolising mutants of cytochrome P450 BM3 (CYP102A1) as biocatalysts for the generation of reactive metabolites. Chem. Biol. Interact. 2008, 171, 96–107.
- Van Vugt-Lussenburg, B.M.A.; Stjernschantz, E.; Lastdrager, J.; Oostenbrink, C.; Vermeulen, N.P.E.; Commandeur, J.N.M. Identification of critical residues in novel drug metabolizing mutants of Cytochrome P450 BM3 using random mutagenesis. J. Med. Chem. 2007, 50, 455–461.
- Budde, M.; Morr, M.; Schmid, R.D.; Urlacher, V.B. Selective Hydroxylation of highly branched fatty acids and their derivatives by CYP102A1 from Bacillus megaterium. ChemBioChem 2006, 7, 789–794.
- Vugt-Lussenburg, B.M.A.; Damsten, M.C.; Maasdijk, D.M.; Vermeulen, N.P.E.; Commandeur, J.N.M. Heterotropic and homotropic cooperativity by a drug-metabolising mutant of cytochrome P450 BM3. Biochem. Biophys. Res. Commun. 2006, 346, 810–818.
- Ryu, S.H.; Park, B.Y.; Kim, S.Y.; Park, S.H.; Jung, H.J.; Park, M.; Park, K.D.; Ahn, T.; Kang, H.S.; Yun, C.H. Regioselective hydroxylation of omeprazole enantiomers by bacterial CYP102A1 mutants. Drug Metab. Dispos. 2014, 42, 1493–1497.
- Kim, D.H.; Ahn, T.; Jung, H.C.; Pan, J.G.; Yun, C.H. Generation of the human metabolite piceatannol from the anticancer-preventive agent resveratrol by bacterial cytochrome P450 BM3. Drug Metab. Dispos. 2009, 37, 932–936.
- Jang, H.H.; Ryu, S.H.; Le, T.K.; Doan, T.T.M.; Nguyen, T.H.H.; Park, K.D.; Yim, D.E.; Kim, D.H.; Kang, C.K.; Ahn, T.; et al. Regioselective C-H hydroxylation of omeprazole Sulfide by Bacillus megaterium CYP102A1 to Produce a Human Metabolite. Biotechnol. Lett. 2017, 39, 105–112.
- Le, T.K.; Jang, H.H.; Nguyen, H.T.H.; Doan, T.T.M.; Lee, G.Y.; Park, K.D.; Ahn, T.; Joung, Y.H.; Kang, H.S.; Yun, C.H. Highly Regioselective hydroxylation of polydatin, a resveratrol glucoside, for one-step synthesis of astringin, a piceatannol glucoside, by P450 BM3. Enzyme Microb. Technol. 2017, 97, 34–42.
- Nguyen, N.A.; Jang, J.; Le, T.K.; Nguyen, T.H.H.; Woo, S.M.; Yoo, S.K.; Lee, Y.J.; Park, K.D.; Yeom, S.J.; Kim, G.J.; et al. Biocatalytic production of a potent inhibitor of adipocyte differentiation from phloretin using engineered CYP102A1. J. Agric. Food Chem. 2020, 68, 6683–6691.
- Nguyen, N.A.; Cao, N.T.; Nguyen, T.H.H.; Le, T.K.; Cha, G.S.; Choi, S.K.; Pan, J.G.; Yeom, S.J.; Kang, H.S.; Yun, C.H. Regioselective hydroxylation of phloretin, a bioactive compound from apples, by human Cytochrome P450 Enzymes. Pharmaceuticals 2020, 13, 330.
- Fredenhagen, A.; Schroer, K.; Schröder, H.; Hoepfner, D.; Ligibel, M.; Porchet Zemp, L.; Radoch, C.; Freund, E.; Meishammer, A. Cladosporin derivatives obtained by biotransformation provide guidance for the focused derivatization of this antimalarial lead compound. ChemBioChem 2019, 20, 650–654.
- Venkataraman, H.; de Beer, S.B.A.; van Bergen, L.A.H.; van Essen, N.; Geerke, D.P.; Vermeulen, N.P.E.; Commandeur, J.N.M. A Single Active Site Mutation Inverts Stereoselectivity of 16-hydroxylation of testosterone catalyzed by engineered cytochrome P450BM3. ChemBioChem 2012, 13, 520–523.
- Reinen, J.; Vredenburg, G.; Klaering, K.; Vermeulen, N.P.E.; Commandeur, J.N.M.; Honing, M.; Vos, J.C. Selective whole-cell biosynthesis of the designer drug metabolites 15- or 16-betahydroxynorethisterone by engineered cytochrome P450 BM3 Mutants. J. Mol. Catal. B Enzym. 2015, 121, 64–74.
- Reinen, J.; Ferman, S.; Vottero, E.; Vermeulen, N.P.E.; Commandeur, J.N.M. Application of a fluorescence-based continuous-flow bioassay to screen for diversity of cytochrome P450 BM3 mutant libraries. J. Biomol. Screen. 2011, 16, 239–250.
- Venkataraman, H.; te Poele, E.M.; Rosłoniec, K.Z.; Vermeulen, N.; Commandeur, J.N.M.; van der Geize, R.; Dijkhuizen, L. Biosynthesis of a steroid metabolite by an engineered Rhodococcus Erythropolis strain expressing a mutant cytochrome P450 BM3 enzyme. Appl. Microbiol. Biotechnol. 2015, 99, 4713–4721.
- Rea, V.; Kolkman, A.J.; Vottero, E.; Stronks, E.J.; Ampt, K.A.M.; Honing, M.; Vermeulen, N.P.E.; Wijmenga, S.S.; Commandeur, J.N.M. Active site substitution A82W improves the regioselectivity of steroid hydroxylation by cytochrome P450 BM3 mutants as rationalized by spin relaxation nuclear magnetic resonance studies. Biochemistry 2012, 51, 750–760.
- Cha, G.S.; Ryu, S.H.; Ahn, T.; Yun, C.H. Regioselective hydroxylation of 17β-estradiol by mutants of CYP102A1 from Bacillus megaterium. Biotechnol. Lett. 2014, 36, 2501–2506.
- Sawayama, A.M.; Chen, M.M.Y.; Kulanthaivel, P.; Kuo, M.-S.; Hemmerle, H.; Arnold, F.H. A panel of cytochrome P450 BM3 variants to produce drug metabolites and diversify lead compounds. Chem. A Eur. J. 2009, 15, 11723–11729.
- Liu, X.; Kong, J.-Q. Steroids hydroxylation catalyzed by the monooxygenase mutant 139-3 from Bacillus megaterium BM3. Acta Pharm. Sin. B 2017, 7, 510–516.
- Marchetti, S.; Schellens, J.H.M. The impact of FDA and EMEA guidelines on drug development in relation to phase 0 trials. Br. J. Cancer 2007, 97, 577–581.
- Food and Drug Administration Center for Drug Evaluation and Research. Safety Testing of Drug Metabolites: Guidance for Industry; FDA: Silver Spring, MD, USA, 2016.
- Kim, D.H.; Kim, K.H.; Kim, D.; Jung, H.C.; Pan, J.G.; Chi, Y.T.; Ahn, T.; Yun, C.H. Oxidation of human cytochrome P450 1A2 substrates by Bacillus megaterium cytochrome P450 BM3. J. Mol. Catal. B Enzym. 2010, 63, 179–187.
- Rentmeister, A.; Brown, T.R.; Snow, C.D.; Carbone, M.N.; Arnold, F.H. Engineered Bacterial Mimics of Human Drug Metabolizing Enzyme CYP2C9. ChemCatChem 2011, 3, 1065–1071.
- Tsotsou, G.E.; Sideri, A.; Goyal, A.; Di Nardo, G.; Gilardi, G. Identification of mutant Asp251Gly/Gln307His of cytochrome P 450 BM3 for the generation of metabolites of diclofenac, ibuprofen and tolbutamide. Chem. A Eur. J. 2012, 18, 3582–3588.
- Di Nardo, G.; Dell’Angelo, V.; Catucci, G.; Sadeghi, S.J.; Gilardi, G. Subtle structural changes in the Asp251Gly/Gln307His P450 BM3 mutant responsible for new activity toward diclofenac, tolbutamide and ibuprofen. Arch. Biochem. Biophys. 2016, 602, 106–115.
- Ren, X.; Yorke, J.A.; Taylor, E.; Zhang, T.; Zhou, W.; Wong, L.L. Drug oxidation by cytochrome P450BM3: Metabolite synthesis and discovering new P450 reaction types. Chem. A Eur. J. 2015, 21, 15039–15047.
- Venkataraman, H.; Verkade-Vreeker, M.C.A.; Capoferri, L.; Geerke, D.P.; Vermeulen, N.P.E.; Commandeur, J.N.M. Application of engineered cytochrome P450 mutants as biocatalysts for the synthesis of benzylic and aromatic metabolites of fenamic acid NSAIDs. Bioorg. Med. Chem. 2014, 22, 5613–5620.
- Butler, C.F.; Peet, C.; McLean, K.J.; Baynham, M.T.; Blankley, R.T.; Fisher, K.; Rigby, S.E.J.; Leys, D.; Voice, M.W.; Munro, A.W. Human P450-like oxidation of diverse proton pump inhibitor drugs by “gatekeeper” mutants of flavocytochrome P450 BM3. Biochem. J. 2014, 460, 247–259.
- Le, T.K.; Cha, G.S.; Jang, H.H.; Nguyen, T.H.H.; Doan, T.T.M.; Lee, Y.J.; Park, K.D.; Shin, Y.; Kim, D.H.; Yun, C.H. Regioselective hydroxylation pathway of tenatoprazole to produce human metabolites by Bacillus megaterium CYP102A1. Process Biochem. 2019, 87, 95–104.
- Richards, L.; Lutz, A.; Chalmers, D.K.; Jarrold, A.; Bowser, T.; Stevens, G.W.; Gras, S.L. Production of metabolites of the anti-cancer drug noscapine Using a P450BM3 mutant library. Biotechnol. Rep. 2019, 24, e00372.
- Koyani, R.D.; Vazquez-Duhalt, R. Enzymatic activation of the emerging drug resveratrol. Appl. Biochem. Biotechnol. 2018, 185, 248–256.
- Luirink, R.A.; Dekker, S.J.; Capoferri, L.; Janssen, L.F.H.; Kuiper, C.L.; Ari, M.E.; Vermeulen, N.P.E.; Vos, J.C.; Commandeur, J.N.M.; Geerke, D.P. A combined computational and experimental study on selective flucloxacillin hydroxylation by cytochrome P450 BM3 variants. J. Inorg. Biochem. 2018, 184, 115–122.
- Kim, D.H.; Kim, K.H.; Kim, D.H.; Liu, K.H.; Jung, H.C.; Pan, J.G.; Yun, C.H. Generation of human metabolites of 7-ethoxycoumarin by bacterial cytochrome P450 BM3. Drug Metab. Dispos. 2008, 36, 2166–2170.
- Otey, C.R.; Bandara, G.; Lalonde, J.; Takahashi, K.; Arnold, F.H. Preparation of human metabolites of propranolol using laboratory-evolved bacterial cytochromes P450. Biotechnol. Bioeng. 2006, 93, 494–499.
- Nguyen, T.; Yeom, S.-J.; Yun, C.-H. Production of a human metabolite of atorvastatin by bacterial CYP102A1 peroxygenase. Appl. Sci. 2021, 11, 603.
- Lechner, A.; Brunk, E.; Keasling, J.D. The need for integrated approaches in metabolic engineering. Cold Spring Harb. Perspect. Biol. 2016, 8, a023903.
- Urlacher, V.B.; Girhard, M. Cytochrome P450 monooxygenases in biotechnology and synthetic biology. Trends Biotechnol. 2019, 37, 882–897.
- Dezvarei, S.; Lee, J.H.Z.; Bell, S.G. Stereoselective hydroxylation of isophorone by variants of the cytochromes P450 CYP102A1 and CYP101A1. Enzyme Microb. Technol. 2018, 111, 29–37.
- Kaluzna, I.; Schmitges, T.; Straatman, H.; Van Tegelen, D.; Mü, M.; Schü, M.; Mink, D. Enabling selective and sustainable p450 oxygenation technology. Production of 4-hydroxy-α-isophorone on kilogram scale. Org. Process Res. Dev. 2016, 20, 814–819.
- Ilie, A.; Harms, K.; Reetz, M.T. P450-catalyzed regio- and stereoselective oxidative hydroxylation of 6-Iodotetralone: Preparative-scale synthesis of a key intermediate for Pd-catalyzed transformations. J. Org. Chem. 2018, 83, 7504–7508.
- Klaus, T.; Seifert, A.; Häbe, T.; Nestl, B.M.; Hauer, B. An Enzyme cascade synthesis of vanillin. Catalysts 2019, 9, 252.
- Dennig, A.; Weingartner, A.M.; Kardashliev, T.; Müller, C.A.; Tassano, E.; Schürmann, M.; Ruff, A.J.; Schwaneberg, U. An enzymatic route to α-tocopherol synthons: Aromatic hydroxylation of pseudocumene and mesitylene with P450 BM3. Chem. A Eur. J. 2017, 23, 17981–17991.
- Weingartner, A.M.; Sauer, D.F.; Dhoke, G.V.; Davari, M.D.; Ruff, A.J.; Schwaneberg, U. A hydroquinone-specific screening system for directed P450 evolution. Appl. Microbiol. Biotechnol. 2018, 102, 9657–9667.
- Munday, S.D.; Dezvarei, S.; Lau, I.C.K.; Bell, S.G. Examination of Selectivity in the Oxidation of Ortho- and Meta-Disubstituted Benzenes by CYP102A1 (P450 Bm3) Variants. ChemCatChem 2017, 9, 2512–2522.
- Carmichael, A.B.; Wong, L.L. Protein engineering of Bacillus megaterium CYP102A1. The oxidation of polycyclic aromatic hydrocarbons. Eur. J. Biochem. 2001, 268, 3117–3125.
- Whitehouse, C.J.C.; Bell, S.G.; Tufton, H.G.; Kenny, R.J.P.; Ogilvie, L.C.I.; Wong, L.L. Evolved CYP102A1 (P450BM3) variants oxidise a range of non-natural substrates and offer new selectivity options. Chem. Commun. 2008, 8, 966–968.
- Holec, C.; Hartrampf, U.; Neufeld, K.; Pietruszka, J. P450 BM3-catalyzed regio- and stereoselective hydroxylation aiming at the synthesis of phthalides and isocoumarins. ChemBioChem 2017, 18, 676–684.
- Wang, Z.J.; Renata, H.; Peck, N.E.; Farwell, C.C.; Coelho, P.S.; Arnold, F.H. Improved Cyclopropanation activity of histidine-ligated Cytochrome P450 enables the enantioselective formal synthesis of levomilnacipran. Angew. Chemie Int. Ed. 2014, 53, 6810–6813.
- Li, Y.; Wong, L.L. Multi-functional oxidase activity of CYP102A1 (P450BM3) in the oxidation of quinolines and tetrahydroquinolines. Angew. Chemie Int. Ed. 2019, 58, 9551–9555.
- Li, Y.; Qin, B.; Li, X.; Tang, J.; Chen, Y.; Zhou, L.; You, S. Selective oxidations of cyperenoic acid by slightly reshaping the binding pocket of cytochrome P450 BM3. ChemCatChem 2018, 10, 559–565.
- Acevedo-Rocha, C.G.; Gamble, C.G.; Lonsdale, R.; Li, A.; Nett, N.; Hoebenreich, S.; Lingnau, J.B.; Wirtz, C.; Fares, C.; Hinrichs, H.; et al. P450-catalyzed regio-and diastereoselective steroid hydroxylation: Efficient directed evolution enabled by mutability Landscaping. ACS Catal. 2018, 8, 3395–3410.
- Chu, L.L.; Pandey, R.P.; Lim, H.N.; Jung, H.J.; Thuan, N.H.; Kim, T.S.; Sohng, J.K. Synthesis of umbelliferone derivatives in Escherichia Coli and their biological activities. J. Biol. Eng. 2017, 11, 1–11.
- Chu, L.L.; Pandey, R.P.; Jung, N.; Jung, H.J.; Kim, E.H.; Sohng, J.K. Hydroxylation of Diverse Flavonoids by CYP450 BM3 variants: Biosynthesis of eriodictyol from naringenin in whole cells and its biological activities. Microb. Cell Fact. 2016, 15, 135.
- Hong, L.L.; Kong, J.Q. Altering the Regioselectivity of Cytochrome P450 BM3 variant M13 toward genistein through protein engineering and variation of reaction conditions. ACS Omega 2020, 5, 32059–32066.
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chemie Int. Ed. 2021, 60, 88–119.
- Chen, W.; Fisher, M.J.; Leung, A.; Cao, Y.; Wong, L.L. Oxidative diversification of steroids by nature-inspired scanning glycine Mutagenesis of P450BM3 (CYP102A1). ACS Catal. 2020, 10, 8334–8343.
- Li, A.; Acevedo-Rocha, C.G.; D’Amore, L.; Chen, J.; Peng, Y.; Garcia-Borràs, M.; Gao, C.; Zhu, J.; Rickerby, H.; Osuna, S.; et al. Regio- and stereoselective steroid hydroxylation at C7 by cytochrome P450 monooxygenase mutants. Angew. Chemie Int. Ed. 2020, 59, 12499–12505.
- Kolev, J.N.; O’Dwyer, K.M.; Jordan, C.T.; Fasan, R. Discovery of potent parthenolide-based antileukemic agents enabled by late-stage P450-mediated C—H functionalization. ACS Chem. Biol. 2014, 9, 164–173.
- Alwaseem, H.; Frisch, B.J.; Fasan, R. Anticancer activity Profiling of Parthenolide Analogs Generated via P450-mediated chemoenzymatic synthesis. Bioorg. Med. Chem. 2018, 26, 1365–1373.
- Zhang, X.; King-Smith, E.; Dong, L.B.; Yang, L.C.; Rudolf, J.D.; Shen, B.; Renata, H. Divergent synthesis of complex diterpenes through a hybrid oxidative approach. Science 2020, 369, 799–806.
- Kitamura, E.; Otomatsu, T.; Maeda, C.; Aoki, Y.; Ota, C.; Misawa, N.; Shindo, K. Production of hydroxlated flavonoids with cytochrome P450 BM3 variant F87V and their antioxidative activities. Biosci. Biotechnol. Biochem. 2013, 77, 1340–1343.
- Ko, S.; Yang, Y.H.; Choi, K.Y.; Kim, B.G. Rational design and directed evolution of CYP102A1 (BM3) for regio-specific hydroxylation of isoflavone. Biotechnol. Bioprocess Eng. 2015, 20, 225–233.
- Wang, J.B.; Ilie, A.; Reetz, M.T. Chemo- and stereoselective cytochrome P450-BM3-catalyzed sulfoxidation of 1-Thiochroman-4-Ones enabled by directed evolution. Adv. Synth. Catal. 2017, 359, 2056–2060.
- Syntrivanis, L.-D.; Wong, L.L.; Robertson, J. Hydroxylation of eleuthoside synthetic Intermediates by P450 BM3 (CYP102A1). Eur. J. Org. Chem. 2018, 2018, 6369–6378.
- Vickers, C.; Backfisch, G.; Oellien, F.; Piel, I.; Lange, U.E.W. Enzymatic Late-stage oxidation of lead compounds with solubilizing biomimetic docking/protecting groups. Chem. A Eur. J. 2018, 24, 17936–17947.

