Most commercially available pharmaceuticals are produced via traditional organic synthesis routes. However, this approach is limited by the scope of traditional chemical catalysts that often yield predictably similar products. This leads to a difficulty in the synthesis of compounds that exhibit enantioselectivity and structural motifs such as specific hydroxyl groups. There is also a commercial interest in reducing manufacturing costs. Due to these factors, efforts are being made to move future pharmaceutical synthesis towards alternative routes of production that focus on sustainability and cost efficiency
[1]. One area of current interest is biosynthetic manufacture, which harnesses bacterial cells and enzymes for metabolite production as an organic synthesis replacement. These alternatives are both relatively cheap and easy to produce. Additionally, enzymes often have the added advantage of exhibiting higher yields and selectivity towards native small molecule substrates. Of particular interest are the cytochrome P450 (P450 or CYP) enzyme superfamily. These heme enzymes are primarily monooxygenases that are found in most living organisms and share similarities in their characteristic ‘P450′ fold and the highly conserved, cysteine thiolate-coordinated heme prosthetic group
[2].
P450s perform oxidations on a diverse range of organic small molecule substrates and have many physiological roles in biosynthetic and detoxification pathways
[3]. A major biotransformation that is typical of the P450 superfamily is the hydroxylation of carbon–hydrogen bonds via the insertion of an oxygen atom, yielding a hydroxylated metabolite and a molecule of water. The metabolism of substrates is achieved by a complex multi-step catalytic cycle via the heme prosthetic group. In this mechanism, the docking of the substrate and the removal of the distal water ligand of the heme iron triggers a series of transient changes in the heme iron state. These changes involve the transfer of two electrons, a proton and the binding of dioxygen. In the canonical catalytic cycle this leads to the eventual abstraction of a substrate proton which then rebounds to produce an oxidized metabolite
[4]. Other P450-catalyzed biotransformations include (but are not limited to) heteroatom oxygenations, dealkylations, epoxidations, ring cleavages, dehydrogenations and sulfoxidations
[3]. Human P450s have numerous physiological functions and are widely known for their role in xenobiotic metabolism, typically adding polar functional groups, facilitating an excretion of the product
[5]. Since the discovery of P450s there has been an interest in utilizing the P450 superfamily as an alternative route for the synthesis of valuable small molecules, as their mechanistic capabilities can yield oxy-metabolites that are laborious to synthesize via traditional organic synthesis routes. The main drug-metabolizing human P450s exhibits promiscuity and wide substrate profiles. However, due to their eukaryotic nature and N-terminal membrane anchor they can be difficult to express in bacterial hosts that are typically used for biocatalytic cascades. Furthermore, their functioning requires the use of a reductase partner to supply the electron necessary for catalysis. In general, human P450s are catalytically slow. Therefore, there is an advantage to be gained from utilizing soluble bacterial P450s as biocatalysts for metabolite production.
P450 BM3
A bacterial P450 that has been explored extensively for metabolite production capabilities is the natural fusion protein, flavocytochrome P450 BM3 (BM3, CYP102A1)
[6][7][8][6,7,8]. BM3 was isolated in the early 1970s from the soil bacterium,
Bacillus megaterium (the third P450 to be extracted from
B. megaterium), and was identified as a fatty acid hydroxylase that did not require the addition of an independent redox partner
[6][7][6,7]. Structural studies reveal that BM3 is a 119.5 kilodalton (kDa) single polypeptide enzyme of 1048 amino acids that forms a soluble dimer in solution. It contains a 55 kDa heme domain that is fused via a short linker region to a 65 kDa reductase domain that houses FAD and FMN flavins
[9][10][9,10]. An interesting observation is that both domains exhibit structural similarities to eukaryotic microsomal P450-containing systems. Specifically, there is the similarity of the reductase domain to human cytochrome P450 reductuase (CPR) and of the heme domain to the CYP2A and CYP4A fatty acid hydroxylase sub-families, while there is a lack of the N-terminal encoded transmembrane anchor
[11][12][11,12]. P450 BM3′s precise function in vivo is not particularly well defined, but it is postulated that it plays a role in the detoxification of xenobiotic lipids produced by plants
[13][14][13,14].
The BM3 heme domain was among the first P450 crystal structures to be published, shedding light on important motifs and residues within the protein (PDB code: 2HPD)
[15]. BM3 exhibits a structural integrity conserved across the P450 superfamily, specifically within the B’/C loop, the C- and E-helices, the E/F loop, the β5-sheet and the I-helix
[16]. The heme domain of BM3 is relatively flexible and stable and contains the heme prosthetic group necessary for catalysis
[17]. BM3 also contains a long, hydrophobic, non-aromatic substrate access channel, which is hypothesized to exist in a state of equilibrium between the substrate-free and substrate-bound forms
[18][19][18,19]. To date, the intact structure of BM3 has yet to be solved to a higher resolution, most likely due to the dynamic nature of the reductase domain.
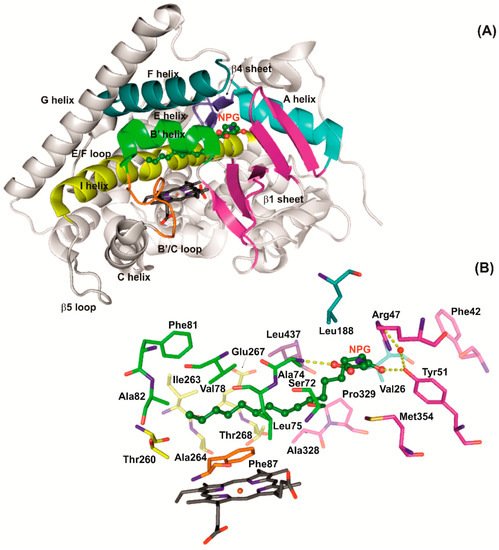
Figure 1 illustrates the secondary structure of the BM3 heme domain in complex with NPG (N-palmitoyl glycine), highlighting the key residues that play roles in the substrate binding.
Figure 1. The secondary structure and active site of NPG-bound WT BM3 heme domain (PDB: code 1JPZ). (A) Secondary structure of WT BM3 with key regions indicated. The heme group is shown as dark grey sticks with the iron in brown and NPG shown as ball and sticks in dark green. Regions that form the active site and substrate access channel are in multi-colors. (B) The substrate access channel and active site of NPG-bound WT BM3. Heme and NPG are indicated as in panel A. All oxygens are in dark blue and nitrogens in red. Key residues involved in substrate binding are indicated in colors corresponding to their secondary structure regions in (A). Polar contacts of NPG with active site waters (red spheres) and residues Arg47, Tyr51 and Ala74 are indicated by yellow dashed lines.
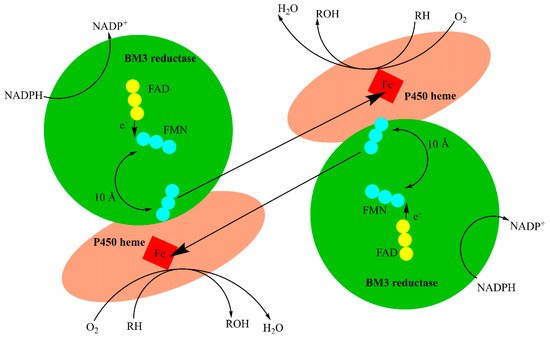
As mentioned above, BM3 catalysis is an electron-dependent process requiring two electrons delivered from the flavin-containing reductase domain that is attached to the heme domain via the linker region. This fusion yields a highly efficient system with significantly increased activity relative to other monooxygenase P450s. This efficiency enhancement is attributed to the redox partner’s proximity to the heme iron. To date BM3 has the highest reported catalytic activity of any known monooxygenase
[20]. Electrons are shuttled in a similar way to the process observed in the microsomal cytochrome P450 reductase (CPR or POR). This transfer starts with the NADPH hydride donation and is followed by a series of FAD and FMN radical oxidations that lead to the electron transfer to the heme domain as a part of the catalytic cycle as shown in
Figure 2 [21][22][21,22]. The studies with inactive heme and reductase domains suggest that the electrons are shuttled in an intermolecular manner from the FMN in the reductase domain to the heme irons in either heme domain
[23]. This rapid electron transfer and malleable active site made BM3 an attractive tool for biotechnological uses.
Figure 2. Hypothetical dimeric BM3 electron transfer. The flavin cofactors, FAD and FMN, facilitate electron transfer from the reductase domain to the heme domain. FAD (yellow circles) transfers electrons to FMN (blue circles, which then undergoes a structural rearrangement to facilitate electron transfer to the heme iron (red square).
There are several key residues in BM3 that play a critical role in the structure and function of both the heme and reductase domains, which over the years have made them attractive sites for mutagenesis. For example, to shift the specificity of BM3 to a structurally diverse range of organic substrates or to investigate their structural and mechanistic properties.
Figure 1 illustrates the interactions of several of these key residues in complex with NPG. Within the heme domain, Arg47 and Tyr51 sit at the mouth of the substrate access channel and form a hydrophilic binding region that holds fatty acid substrates in place via the carboxylate group
[24]. Phe42 provides a ‘cap’ to the substrate access channel in the substrate-bound conformation that is important for catalytic efficiency
[15][25][15,25]. Phe87 is located near the heme iron in the substrate access channel and plays a role in displacing the axial water molecule which triggers the active site reorganization. It has been demonstrated that Phe87 is a residue of great importance in controlling selectivity and specificity
[24]. Other active site residues that have been identified as desirable mutagenesis sites for substrate diversification are Ser72, Val78, Ala82 and Leu188, which play key roles in active site conformations
[26]. Glu267 and Thr268 are conserved residues near the kink region of the I-helix (
Figure 1, in yellow) and play key roles in proton delivery, oxygen activation and the stabilization of several catalytic cycle intermediates
[27][28][27,28]. Ala264 is a residue on the I-helix that is postulated to draw the axially coordinated water ligand from the binding site upon the I-helix rearrangement which is typical of substrate binding
[29]. Whereas the highly conserved Phe393 residue plays a key role in oxygen binding
[30][31][30,31].
2. Wild Type BM3 and Pharmaceutical Metabolism
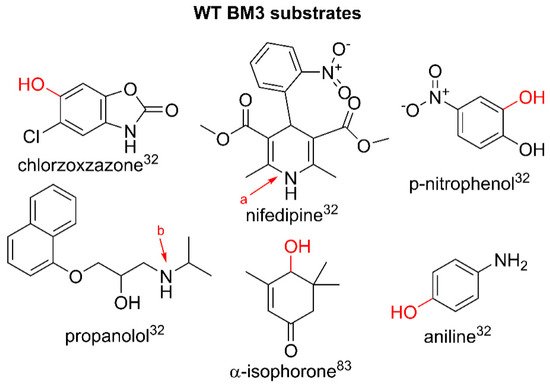
Whilst the wild-type enzyme has a comparatively narrow active site channel and reluctantly binds small molecules that deviate from its hypothesized mid-chain-length, fatty-acid native substrates, there are examples that show the metabolism of drug-like precursors and pharmaceuticals. The studies on WT BM3 have shown that the enzyme can accept substrates metabolized by human P450 isoforms, including catalyzing the hydroxylation of structurally diverse compounds such as chlorzoxazone, aniline and
p-nitrophenol, the N-dealkylation of propranolol and the dehydrogenation of nifedipine. The V
max for all drugs was either comparable or higher than their human P450 counterparts and it was found that WT BM3 exhibited product profiles similar to human CYP3A4 and CYP2E1
[32]. Other examples of WT BM3 metabolizing drug-like compounds show a relatively low activity in comparison to engineered variants designed through iterative mutagenesis studies, including the vitamin A precursor β-ionone, 12-p-nitrophenoxy-carboxylic acid and propylbenzene
[33][34][35][33,34,35].
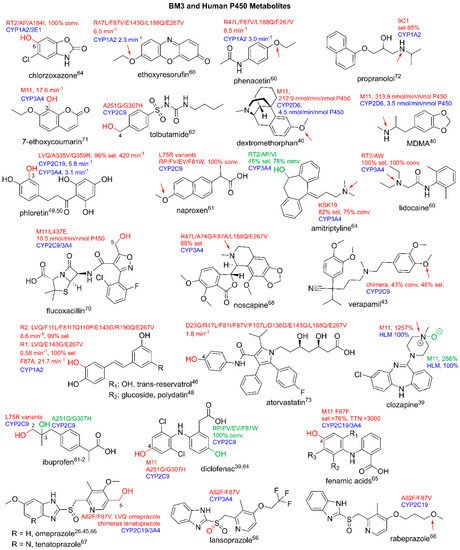
Figure 3 illustrates the modifications on selected drug substrates by WT BM3.
Figure 3. Substrates metabolized by WT BM3. Parent compound structures are in black. Sites of modification are indicated in red: a. aromatization of nifedipine to yield dehydronifedipine, b. dealkylation of propranolol. Literature references are in superscript.
3. Engineering BM3 for Drug Metabolite Production
Due to the structural similarity of the BM3 heme domain to the human drug-metabolizing P450s, coupled with the enzyme’s high catalytic efficiency and malleable active site, this domain is an attractive target for mutagenesis studies with the aim of producing pharmaceutical metabolites. The following section aims to give a comprehensive overview of BM3 engineering for the production of valuable drug metabolites.
3.1. Key Promiscuous BM3 Variants and Library Screening
In early studies, BM3 was targeted as a potential bacterial homologue to many human drug-metabolizing P450s with the aim of shifting the specificity for the production of large quantities of metabolites in an efficient and cost-effective manner. The iterative mutagenesis studies on key active site residues have led to the identification of BM3 variants that exhibit substrate promiscuity. Some of the earliest directed evolution studies on BM3 in Frances Arnold’s group led to the design of the promiscuous variants, 9-10A and 139-3 (see
Table 1 for details), which have since been carried forward in subsequent studies on pharmaceutical metabolite production for classes such as steroids, alkaloids, and monosaccharides
[36][37][38][36,37,38]. Promiscuous variants were designed based on the triple mutant LVQ in Nico Vermeulen’s group through iterations of error-prone PCR and were shown to exhibit activities up to 70-fold higher than human P450s for clozapine, diclofenac, and acetaminophen
[39]. M11 also showed a 200-fold increase in the turnover rate towards dextromethorphan and 3,4-methylenedioxymethylamphetamine compared to WT BM3 and a 90-fold increase compared to human CYP2D6
[40]. The triple mutant GVQ, identified by saturation mutagenesis studies in the Urlacher and Commandeur groups, was found to hydroxylate the omega positions of branched fatty acid derivatives. Further studies demonstrated the ability of GVQ to metabolize structurally diverse human P450 substrates
[41][42][41,42]. In the above examples of promiscuous variants, there is a clear trend of the introduction of the same or similar point mutations which expand BM3′s catalytic repertoire, including: the neutralization of the polar binding mouth of the active site granting access to non-polar substrates (R47L/Y51F), the addition of polar residues to counteract an overly non-polar environment (L188Q), the removal of Phe87 steric hindrance (F87A/V/L/G) and active site reorganizations (A82L/F, A74E, V78L/F). These residues are often utilized as initial mutagenesis targets in studies aiming to shift the specificity of BM3 to non-natural substrates.
Table 1. Promiscuous BM3 variants and their corresponding drug substrates. Asterisks indicate where further point mutations were introduced for metabolism.
| BM3 Variant |
Point Mutations |
Substrates |
| LVQ [39] |
R47L/F87V/L188Q |
clozapine [39], diclofenac [39], acetaminophen [39], omeprazole [43][45], reservatrol * [44][46], polydatin [ |
3.4. Utilizing BM3 for Production of Emerging Pharmaceuticals
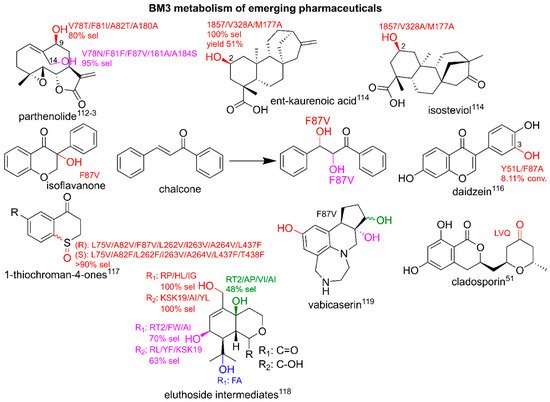
The research into the biosynthetic production of pharmaceuticals has led to harnessing and engineering enzymes from natural product cascades for a generation of emerging pharmaceuticals to explore chemical space that is difficult to reach by means of chemical synthesis
[93][108]. As detailed previously, the catalytic repertoire of BM3 extends beyond what is typically available in traditional synthesis routes, and there are many examples of the production of analogues resulting from BM3-mediated oxidation modifications not previously observed. This section gives an overview of the utilization of BM3 for the production of emerging pharmaceuticals and is summarized in
Figure 7.
Figure 7. BM3-mediated metabolism of emerging pharmaceuticals. Parent compounds are in black. Modifications are in red, pink, green and blue. Conv, conversion; sel, selectivity. Literature references are in superscript.
Previously, the production of BM3 and human steroid metabolites was discussed. There were also extensive studies into the hydroxylation of steroid positions that were difficult to reach for late-stage diversification or as molecular handles. The variant, 139-3 (see
Table 1), hydroxylates androstenedione at the challenging 1α-position. This interesting reaction is postulated to be caused by Arg379 which is thought to play a role in active site orientation
[58]. Steroidal C6β- and C7β-alcohols are shown to display neuroprotective and anti-inflammatory properties but are difficult to chemically oxidate. Several variants are shown to hydroxylate at these positions with high selectivities
[94][95][109,110].
Table 2 gives an overview of BM3-mediated steroid hydroxylation.
Table 2. BM3-mediated steroid metabolism. Only sites of modification are identified. * Indicates human CYP3A metabolite. Conv, conversion; sel, selectivity; ee, enantiomeric excess.
| Steroid |
Structure |
OH-
Position |
Variant |
Parameters |
| 45 | ] | [46][47,48], phloretin * [47][48][49, |
| Testosterone |
 | 50], cladosporin [49][51] |
| 2β * |
RLYF/KSK19 * | [ | 64] |
100% conv, 61% sel |
M01 [39] |
LVQ/E267V/G415S/G1049E |
| 1β |
H171L/Q307H/N319Y/F87A/T260G/P329G/A330W [94][109] | clozapine [39], diclofenac [39], acetaminophen [39], testosterone * [50][52], noresthisterone * [51][53], methoxyresorufin [52][44], ethoxyresorufin [52][44] |
| 71% sel, 76% conv |
M02 [39] |
LVQ/L86I/N319T/A964V |
clozapine [39] |
| 7β |
R47W/S72W/F77Y/V78L/F81I/A82L/T88S/M177T/M185Q/L188Q/I209T [95][110] | , diclofenac [39], acetaminophen [39], norandrostenedione [53][54], methoxyresorufin [52][44], ethoxyresorufin [52][44], buspirone [52][44], amitriptyline [52][44], aripiprazole [52][44] |
| 90% sel |
M05 [39] |
LVQ/F81I/E267V/G415S |
clozapine [39], diclofenac [39], acetaminophen [39], methoxyresorufin [52 |
| 15β * |
KSK19/FV/QP [64] | ][44], ethoxyresorufin [52][44] |
| 96% sel, 83% conv |
M11 [39] |
LVQ/E64G/F81I/E143G/E267V/G415S |
clozapine [39], diclofenac [39], acetaminophen [39], Testosterone * [50][54][52,55], norethisterone * [51][54][53,55], dextromethorphan [40], nifedipine [40], midazolam [40], 3,4-methylenedioxymethylamphetamine [40], estradiol [14][55][14,56], methoxyresorufin [52][44], ethoxyresorufin [52][44], buspirone [52][44], duloxetine [52][44], thioridazine [52][44], ondansetron [52][44], imatinib [52][44], omeprazole [52][44], rosiglitazone [52][44], paroxetine [52][44], norethisterone [52][44], resveratrol [52][44], dihydrobenzophenone [52][44], 17α-ethinylestradiol [52][44], bisphenol A [52][44], diethylstilbestrol [52][44], hexestrol [52][44], estriol [52][44], benzophenone-3 [52][44], aldrin [52][44], testosterone [52][44], tramadol [52][44], imidazole [52][44], ketoconazole [52][44] |
| 9-10A [36] |
R47C/V78A/K94I/P142S/T175I/A184V/F205C
/S226R/H236Q/E252G/R255S/A290V/L353V |
verapamil [56][43], astemizole [56][43], LY294002 [56][43] |
| 139-3 [38] |
V78A/H138Y/T175I/V178I/A184V/
H236Q/
E252G/R255S/A290V/A295T/L353V |
androstenedione [57] |
| GVQ [41] |
A74G/F87V/L188Q |
testosterone [42], amodiaquine [42], dextromethorphan [42], acetaminophen [42], 3,4-methylenedioxymethylamphetamine [42] |
Following the above studies, BM3 libraries were designed with the aim of reaching the promiscuity of the major human drug-metabolizing P450s. One of the first reported libraries was designed by the Arnold group and consisted of point mutations and homologue chimeras. Several variants produced metabolites for verapamil, astemizole and LY294002. A trend in variants evolved from 9-10A and point mutations at Phe87, Ala82 and Val78 exhibited the highest promiscuity and conversion rate. Bulkier substitutions at Ala82 and Phe87 displayed higher conversions but decreased metabolite selectivity
[36][56][36,43]. A library based on M11 was tested for the O-dealkylation activity of allyloxy-resorufins, with active variants carried forward for affinity testing against several structurally diverse drugs. The variants displayed differing degrees of affinity and selectivity to every drug, with key metabolites produced for buspirone
[40][52][40,44].
Table 1 gives an overview of the promiscuous variants and their corresponding substrates.
3.2. Production of Human P450 Metabolites
Health governing bodies legally require a full drug metabolism and pharmacokinetic profile of lead drug candidates for progression to phase I clinical trials
[58]. This profile requires the full structural elucidation of drug metabolites produced at 10% or more of the level of the parent compound, as well as the determination of the in vivo effects
[59]. Due to the relatively slow turnover rates of human P450s and the challenges that arise with expressing and purifying membrane-bound proteins, it can be a laborious task to produce metabolites in an easy and cost-efficient manner. BM3′s catalytic similarity to major human P450 drug-metabolizing isoforms and its relative ease of use has made the enzyme an attractive target for mutagenesis studies aiming to shift specificity towards the production of human drug metabolites.
Previously, key variants were described that were designed for the expansion of BM3′s catalytic repertoire for the metabolism of structurally diverse pharmaceuticals. With this in mind, there have been several studies which attempted to design BM3 variants that ‘mimic’ the action of major human drug-metabolizing P450s. The CYP1A2 substrates, phenacetin, ethoxyresorufin and methoxyresorufin, were selectively metabolized into the human metabolites, acetaminophen and resorufin, respectively, by BM3 variants containing combinations of mutations R47L, L86I, F87V and L188Q with an up to three-fold increase in metabolism in comparison to CYP1A2
[60]. The variants containing L75R increase the B’-helix flexibility and BM3’s ability to accept the typically acidic substrates of CYP2C9 and yield human metabolites of ibuprofen and naproxen
[61]. A251G/Q307H also mimics the actions of CYP2C9, selectively producing the human metabolites, 4-hydroxydiclofenac, 2-hydroxyibuprofen and 4-hydroxytolbutamide. Structural studies revealed that A251G breaks a salt bridge between Asp251 and Lys221, increasing flexibility and triggering structural rearrangements leading to the rotation of Phe87
[62][63][62,63]. A panel of variants that aimed to mimic several isoforms of human drug-metabolizing P450s was designed that led to the production of human drug metabolites for the isoforms CYP2C9 (diclofenac and naproxen); CYP1A2 and CYP2E1 (chlorzoxazone); and CYP3A4 (testosterone, lidocaine and amitriptyline)
[64]. Whilst the above variants have the potential to be used as an effective alternative tool for human metabolite production, there are limitations for their use as a replacement for human P450s as they may not exhibit the same metabolic profile. However, their promiscuity can be utilized for the production of human drug metabolites that have been previously identified as crucial for pre-clinical testing.
BM3 variants are shown to target many drugs from the non-steroidal, anti-inflammatory drug class (NSAIDs), for which human metabolites of naproxen, ibuprofen and diclofenac were identified
[39][61][39,61]. Further studies showed that CYP2C9 and CYP1A2 metabolites of fenamic acids were produced with a high selectivity by BM3 variants and exhibited higher turnover rates in comparison to human liver microsomes
[65]. Several drugs in the proton pump inhibitor class are metabolized by BM3 variants, yielding human metabolites. The ‘gatekeeper’ variant, A82F/F87V, for which A82F ‘locks’ the active site in an open conformation and F87V removes a steric hindrance from the active site, produces human metabolites of omeprazole
[26][43][66][26,45,66]. Other proton pump inhibitor metabolites are illustrated in
Figure 4 [66][67][66,67].
Figure 4. Human P450 and BM3 metabolites. Parent compounds are in black. Sites of modifications are in red and green. Human P450 isoforms are in blue. All arrows indicate the site of N- or O-dealkylation. Conv stands for conversion, Sel stands for selectivity. Rates are in min
−1, total turnover number (TTN), nmol/min/nmol P450 and relative percentages. Literature references are in superscript.
Steroids are a major pharmaceutical class that are mainly metabolized by CYP3A isoforms. There are many examples in the literature of the production of human steroid metabolites by variants of BM3. The mutations of interest include S72I, which is highly enantioselective for α-hydroxylations, and A82W, which appears to increase P450 coupling efficiency
[39][40][50][51][54][55][39,40,52,53,55,56].
Figure 5 illustrates in detail the metabolism of steroids to produce both human and non-human metabolites. There are many more examples of human metabolite production by BM3 variants, which include, but are not limited to, anti-cancer agents such as noscapine, beta blockers such as propranolol and antibiotics such as flucloxacillin
[47][48][68][69][70][71][72][73][49,50,68,69,70,71,72,73]. BM3-derived human metabolites are illustrated in
Figure 4.
Figure 5. BM3-mediated pharmaceutical precursor production. Parent compounds are in black. Modifications are in red, blue and green with related variants that target the compounds. Conv, conversion; sel, selectivity; ee, enantiomeric excess. Literature references are in superscript.
The Arnold group designed some of the first reported BM3 chimeras, where the BM3 heme domain was combined with the homologues, CYP102A2 and CYP102A3, as part of a promiscuous library which aimed to expand the catalytic repertoire for the metabolism of structurally diverse compounds
[56][43]. Attempts have also been made to harness the BM3 reductase domain to facilitate the rapid electron transfer to typically slow drug-metabolizing human P450s for the improved production of human metabolites. Constructs have been designed that link the reductase domain to human P450s for the creation of a self-sufficient, chimeric, human–bacterial, P450-containing system.
3.3. Production of Clinically Relevant Pharmaceuticals and Related Precursors
3.3.1. Pharmaceutical Intermediate and Final Product Production
In current research there have been considerable efforts made towards pushing pharmaceutical production, away from traditional organic synthesis routes and towards more economically and environmentally sustainable biosynthesis pathways. There have been several examples of biosynthetic routes in recent years where enzymes were utilized for the production of valuable precursors and final products due to their relatively high yields, low carbon footprint and ability to modify functional groups that were laborious to synthesize chemically
[74][81]. The presence of P450s in many natural product cascades led to the superfamily being of particular interest in the design of engineered biosynthetic pharmaceutical cascades due to their selectivity and their ability to accept small organic compounds and to perform selective hydroxylations which could be used as a molecular handle or for late-stage functionalization
[75][82].
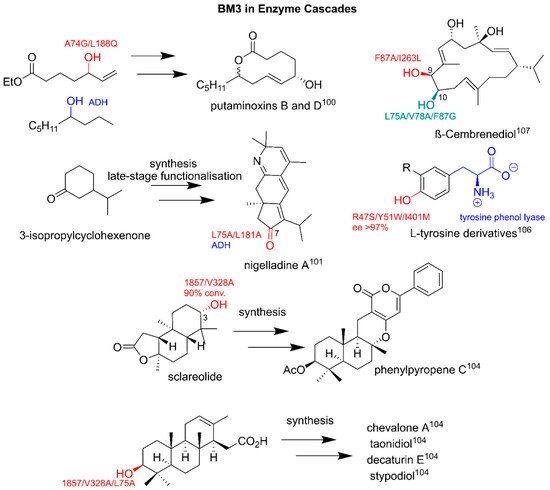
There are several key examples of the BM3-mediated biotransformation of pharmaceutical intermediates. The studies that target alkane-moieties, which are typically cyclohexane derivatives, are of particular interest as specific hydroxylations can yield crucial pharmaceutical precursors. The GVQ variant selectively metabolizes branched fatty acids at omega positions forming precursors to macrolide antibiotics
[41]. WT BM3 stereoselectively hydroxylates α-isophorone to the vitamin E intermediate, 4-hydroxy-α-isophorone, on a kilogram scale (detailed later in this review). Further studies showed that the F87W/Y86F/L244A/V247L modification improved the selectivity by >98%
[76][77][83,84]. The metabolite
(R)-4-hydroxy-iodotetralone, a key intermediate used in palladium-mediated arylations and carbonylations, was produced by F87V at a 99% selectivity
[78][85]. The carotene precursor β-ionone can also be selectively hydroxylated by A74E/F87V/P386S to 4-hydroxy-β-ionone
[35].
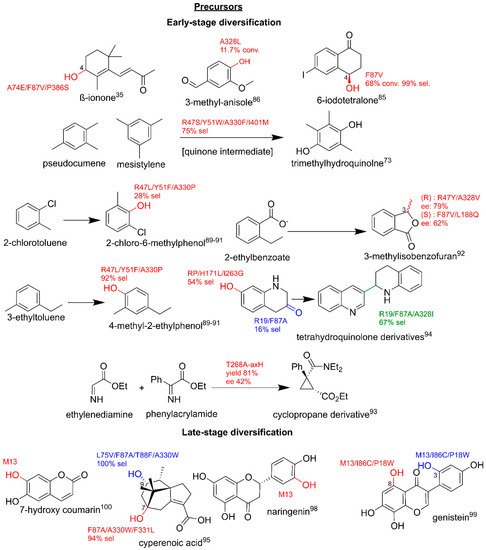
BM3 variants have also been shown to target a range of benzene derivatives. The production of vanillin from 3-methylanisole was achieved in a whole-cell
E. coli system via a three-step cascade using the BM3 variant A328L combined with mutated vanillyl alcohol oxidase, albeit at a low conversion of 11.7% and a yield of 1.1%
[79][86]. WT BM3 and the selected variants also selectively hydroxylate the methyl-substituted benzene derivatives, pseudocumene and mesitylene, to the phenolic building blocks used in the α-tocopherol synthesis via quinolone intermediates. For example, the introduction of the point mutation A330F increased the selectivity towards metabolite trimethylhydroquinolone to 75%
[80][81][87,88]. The selective hydroxylation of various halogenated alkyl benzene derivatives by R47L/Y51F/A330P yields key intermediates such as 2-chlorotoluene and 4-methyl-2-ethylphenol with turnovers of 500 to 1600 min
−1. Interestingly, ortho-substituted toluenes displayed group migrations of the methyl group to meta-positions due to a hypothesized epoxidation, ring opening and migration
[82][83][84][89,90,91]. The stereoselective hydroxylation of 2-alkylated benzoic acid esters to produce phthalide (fungicide intermediates) and isocoumarin (serine protease inhibitors) derivatives was trialed on a preparative scale, which led to the hydroxylation of 2-ethylbenzoate to enantiomers of 3-methylisobenzofuran
[85][92].
The cyclopropanation of phenylacrylamide is the first step in a semi-synthetic route to the antidepressant levomilinacipran. This cyclopropanation reaction was achieved by replacing the axially heme iron coordinated cysteine with histidine (C400H) and by the introduction of the point mutation T268A. In this variant the enzyme no longer proceeds through the canonical mechanism utilizing an iron-oxo intermediate described earlier, but now via a carbene transfer route
[86][93]. A 48-variant library was screened for the production of tetrahydroquinolone derivatives, yielding diverse products with varying degrees of selectivity and functional group modifications. The specific mutations appeared to control the type of biotransformation and led to the expansion beyond BM3’s typical hydroxylations, including A328L which played a role in the metabolite dimer formation. F87A/V, A328I and I263G favored C5 and C6 oxidations and also promoted metabolite dimer formation, with F87 variants displaying activity towards aromatization. The range of biotransformations performed by different variants in this study highlighted BM3’s role as a selective multi-functional oxidase
[87][94].
There are also key examples of utilizing BM3 variants for selective, late-stage oxy-functionalization to yield clinically relevant pharmaceuticals. A late-stage hydroxylation by P450s is advantageous for the addition of functional groups that are chemically laborious to synthesize and removes the need for protecting groups on typically reactive hydroxyl groups. The preparative scale production of cyperenoic acid metabolites gives high yields and high selectivity for products that display antiangiogenic-promoting effects
[88][95]. Glucocorticosteroids are typically steroid-derived C16 alcohols, which require selective hydroxylation; the combinations of BM3 variants at R47, Y51 and F87, displayed selectivities of above 90% (
Figure 6)
[89][96]. M13 variant was utilized for the specific hydroxylations to yield derivatives of existing pharmaceuticals that displayed enhanced medicinal properties
[71]. This variant was also employed for the production of the anticancer and antibacterial agent esculetin on a preparative 3L scale, as well as for synthesizing the more potent flavonoid antibacterial and anticancer agent, eriodictyol. The additional mutations, I86C/P18W, led to the production of 3′-hydroxygenistein and 8-hydroxygenestein, which displayed more potent pharmaceutical effects than the parent compound and the angiogenesis inhibitor genistein
[90][91][92][97,98,99].
Figure 6. BM3 in enzyme cascades. Parent compounds are in black. BM3-related modifications are in red and green. Other enzymes are in blue. Conv, conversion; sel, selectivity; ee, enantiomeric excess. Literature references are in superscript.
| 16α * |
| M11/V87I/S72I |
| [ |
| 50 | ][52]
M01/A82W/S72I [50][52] |
90% ee
95% ee |
| 16β * |
M01/A82W [50][52]
M11/V87I [50][52] |
100% ee
100% ee |
| Estradiol |
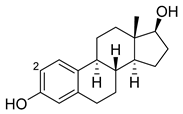 |
2 |
M11 [55][56] |
47 min−1 |
| Norandrostenedione |
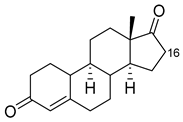 |
16β * |
M02 [53][54] |
95% sel |
| Nandrolone |
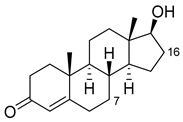 |
7β |
R47W/S72W/F77Y/V78L/F81I/A82L/T88S/M177T/M185Q/L188Q/I209T [95][110] |
75% sel |
| 16α |
R47L/S72I/A82F/F87I/L188C/A330W [89][96] |
98% sel |
| 16β |
R47W/A82W/F87V/L181Q [89][96] |
90% sel |
| Norethindrone |
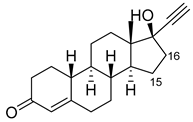 |
15β * |
M11/A82W/V87A [51][53] |
100% sel |
| 16β * |
M01/A82W [51][53] |
96% sel |
| Boldenone |
 |
16α |
R47L/Y51W/S72I/A82W/F87I/L181C [89][96] |
97% sel |
| 16β |
R47W/A82W/F87V/L181Q [89][96] |
72% sel |
| Androstenedione |
 |
1α |
139-3 [57] |
|
| 6β |
H171L/Q307H/N319Y/F87V/I263G [94][109] |
78% sel, 29% conv |
| 7β |
R47W/S72W/F77Y/V78L/F81I/A82L/T88S/M177T/M185Q/L188Q/I209T [95][110]
R47L/Y51F/H171L/Q307H/N319Y/ F87A/A184I/T260G/A328G [94][109] |
90% sel
81% sel, 94% conv |
| 16α |
R47W/Y51W/S72I/A82F/F87I/L181C [89][96] |
95% sel |
| 16β |
R47W/Y51H/A82W/F87V/L181Q [89][96] |
100% sel |
Terpenoids are attractive targets for BM3 diversification. The sesquiterpene lactone parthenolide was previously shown to possess anti-cancer activity but requires the improvement of pharmacokinetic properties. The BM3-mediated addition of hydroxyl groups as molecular handles at the C9- and C14- allowed the addition of the aromatic functional groups and led to the discovery of potent parthenolide-based antileukemic agents
[96][97][111,112]. Studies also aimed to diversify meroterpenoids; selective hydroxylations at the C2-positions of
ent-kaurenoic acid and a derivative of isosteviol led to the production of cochleareine and spiramilactone C derivatives
[98][113].
The diversification of the flavonoid class of pharmaceuticals was explored using variants of BM3 as they displayed a wide range of medicinal properties. The flavanone derivative, s chalcone and isoflavone, were hydroxylated in multiple positions by F87V, with dihydroxyflavanone derivatives displaying the highest IC
50 values for antioxidant activity via testing the lipid peroxidation in rat brain homogenate
[99][114]. Further studies showed that the antioxidant daidzein was turned over by Y51L/F87A to 3-hydroxydaidzein, a compound with more potent effects
[100][115].
The diversification of building blocks for pharmaceuticals is also of interest as the molecular handle of the hydroxyl groups can lead to a wide range of new analogues. The sulfoxidation of 1-thiochroman-4-ones can lead to building blocks of pharmaceutical interest with potential anti-cancer properties. BM3 was engineered for
(S)- and
(R)-sulfoxidation as a replacement to chiral metal catalysts with high selectivities for specific variants displayed
[101][116]. Other key examples of the BM3-mediated diversification of pharmaceuticals include the stereoselective hydroxylation of eleuthoside intermediates, related to the emerging anti-cancer agent, eleuthorobin.
From a panel of BM3 variants including GVQ, the hydroxylation of cyclohexane-based moieties produced a range of oxidized metabolites with high conversion rates and varying degrees of selectivity for several positions. Phe87 variants displayed the ability to further oxidize alcohols to aldehydes with a high regioselectivity and preparative scale reactions showed the ability of BM3 to produce lead drug candidates under scaled-up conditions
[102][117]. The late-stage functionalization of the antimalarial compound cladosporin by LVQ led to the production of ketone derivatives that expressed some degree of potency in the micromolar range against
P. falciparum [49][51]. Late-stage oxidations can present a challenge with selectivity and the production of unwanted by-products. An attempt to solve this issue was trialed with vabicaserin with the addition of a protecting group to ‘steer’ the oxidation site of BM3 variants; an analogue was metabolized by F87V to yield several metabolites with hydroxylations at various positions before the cleavage of the protecting groups
[103][118].