 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Janusz Blasiak | + 9420 word(s) | 9420 | 2021-10-25 03:59:11 |
Video Upload Options
Replication timing (RT) is a cellular program to coordinate initiation of DNA replication in all origins within the genome. RIF1 (replication timing regulatory factor 1) is a master regulator of RT in human cells. This role of RIF1 is associated with binding G4-quadruplexes and changes in 3D chromatin that may suppress origin activation over a long distance.
1. Introduction
2. RIF1—The Gene and the Protein
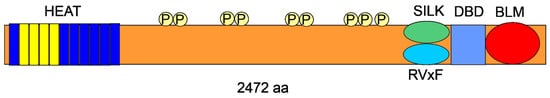
3. Replication Timing
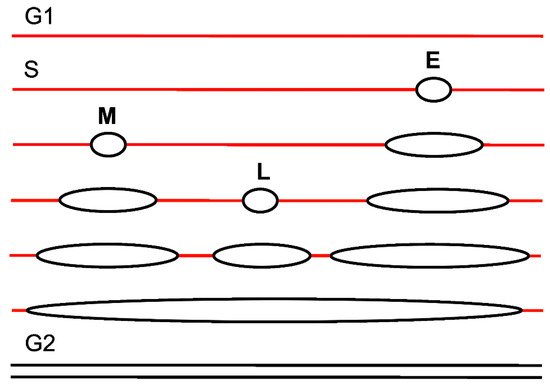
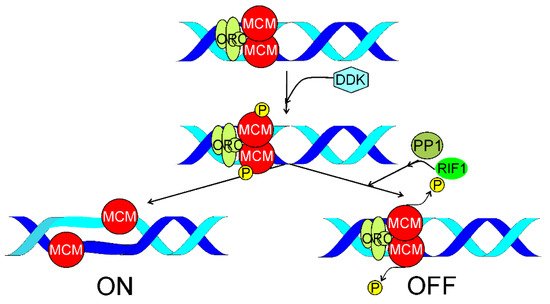
4. RIF1 in Replication Timing
4.1. Organizing the 3D Structure of Chromatin
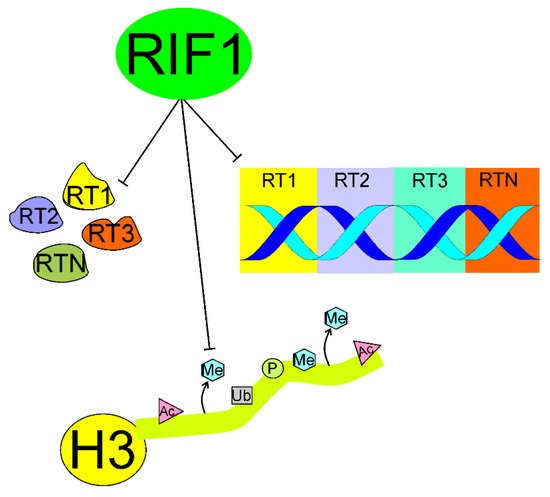

4.2. Interaction with G4-Quadruplexes
DNA double-strand breaks strongly contribute to genomic instability and are potentially lethal if not repaired or misrepaired. DSBs are primarily sensed by the MRN (MRX in yeast) (MRE11 (MRE11 homolog, double strand break repair nuclease)/RAD50 (RAD50 double strand break repair protein)/NBS1 (nibrin, Nijmegen breakage syndrome 1) complex (reviewed in [82]). MRN triggers DDR by the interaction with ataxia telangiectasia mutated (ATM, ATM serine/threonine kinase), and ATR (ATM-and-RAD3-related, ATR serine/threonine kinase) proteins, causing the DSB signaling, signal transduction and effectors action, including DNA repair. As mentioned, in response to DSBs, the cell has evolved three general DSBR pathways: HRR, NHEJ and SSA with several variants to target DSBs (reviewed in [4]). The mechanisms of choice of DSBR depend on the structure of DNA broken ends, cell type, cell cycle phase/chromatin organization and several other factors and are not completely clear [83]. Accumulating evidence suggests that the choice of DSBR pathway in replicating cells differs from that in their non-replicating counterpart (reviewed in [5]). Here, we limit our considerations to HRR and NHEJ as they are much more common than SSA [84].
Although the choice between HRR and NHEJ is temporally regulated by the cell cycle progression, end resection, a prerequisite of HRR rarely occurring in NHEJ, may be a major operational factor deciding about the choice of DSBR pathway at a DSB with blunt ends. Also, the structure of DSB-associated ends may decide about DSBR pathway. One-ended DSBs cannot be repaired by NHEJ or SSA as there is no other end to join. As estimated by Shibata, in G2 about 70% of all two-ended DSBs are repaired by NHEJ and remaining 30% – by HRR [85].
Uncapped telomeres can be recognized as one-sided DSBs and proceeded by DDR resulting in fusion chromosomes, containing fusion genes that frequently act as oncogenes (reviewed in [86]). Therefore, DDR factors that constitutively serve to protect telomeres, may induce their fusion. The nucleotide excision repair endonuclease ERCC1 (XRCC1, X-ray repair cross complementing 1)/XPF (XPF, DNA damage recognition and repair factor) and NHEJ factors Ku70 (X-ray repair cross complementing 5)/80 and DNA-PKcs (protein kinase, DNA-activated, catalytic subunit) are examples of DDR components that may be involved in such ambiguous action [87,88].
It was shown that yeast Rif1 negatively regulated telomere length through its conserved HEAT repeats independently of its role in RT [89,90]. However, as underlined above, the role of human RIF1 in telomere maintenance is clearly distinct from that of yeast Rif1. Human RIF1 does not bind telomere capping complex and join telomeres only if they uncapped, broken or critically short [20,91,92]. RIF1 binds at dysfunctional mammalian telomeres to recover them with its DSBR-related activity rather than telomere-specific functions. However, Adams and McLaren showed that mouse RIF1 highly expressed in primordial germ cells and embryo-derived pluripotent stem cell lines directly interacted with the telomere-associated protein TRF2 and could be crosslinked to telomeric repeat DNA in mouse embryonic stem cells [93]. These authors also showed that RIF1 assisted to limit the expression of the ZSCAN4 (zinc-finger and SCAN domain-containing 4) gene, whose product supports a recombination-related mechanism of telomere-elongation. Therefore, RIF1 can be involved in DSBR-independent telomere maintenance in the germline and early mouse development. Further works are needed to definitely determine the role of mammalian RIF1 in telomere maintenance and clarify the reason of difference between that role and its yeast counterpart.
TRF1 (TERF1, telomeric repeat binding factor 1) is a protein involved in telomere maintenance [94]. It was shown that a fraction of TRF1 may exist in a free form, not bound to telomeric chromatin, when phosphorylated at T371 by CDK1 [95]. McKerlie et al. showed that (pT371)TRF1 was recruited to DSB sites and formed DSB-induced foci in response to DSB-inducing factors [96]. They also showed that depletion of either TP53BP1 (53BP1, tumor protein P53 binding protein 1) or RIF1 stimulated the recruitment of (pT371)TRF1 to DSB sites. Therefore, RIF1 may, independently of TP53BP1, inhibit the binding of an important DDR protein.
5.1. Essential role of TP53BP1
In most effects related to DSBR, RIF1 associates with TP53BP1 [97]. TP53BP1 is a chromatin-binding DDR protein that is recruited to DSB sites via the interaction of its Tudor domain with dimethylated lysine 20 of histone H4 (H4K20me2) [98]. TP53BP1 regulates the choice of DSBR pathway by inhibiting resection of DNA ends by nucleases and consequently suppressing HRR [99].
Several mechanisms may underline regulatory action of TP53BP1, including enforcing a chromatin barrier against nucleases [100]. CBP (CREB binding protein)-mediated acetylation of K1626/1628 in the ubiquitylation-dependent recruitment (UDR) motif was reported to impair the interaction of TP53BP1 with nucleosomes, and consequently block the recruitment of TP53BP1 and its downstream factors RIF1 and PTIP (PAX interacting protein 1) to DSB sites [101,102]. The acetylation of TP53BP1 was tightly regulated by histone deacetylase 2 (HDAC2) to sustain the balance between the HRR and NHEJ pathways.
Silverman et al. observed that human RIF1 accumulated at DSB sites and formed foci colocalizing with DDR factors [18]. They did not observe an accumulation of RIF1 at functional telomeres. Formation of the DDR foci required the function of ATM, but not ATR. RIF1 involvement in the DDR foci was strongly dependent on TP53BP1. However, the RIF1 response was not related to several other important DDR proteins, including CHK2 (checkpoint kinase 2), MRE11, NBS1 and BRCA1 (BRCA1 DNA repair associated). The intra-S checkpoints slow down DNA synthesis to give the cell more time to repair DNA damage and RIF1 functioned in one of the MRN-mediated and ATM-regulated intra-S-phase checkpoints. The authors concluded that RIF1 had a unique pattern of regulation at DSB sites depending primarily on ATM and TP53BP1.
Wang et al. demonstrated that RIF1 downregulation caused defective HRR in cancer cells making them more sensitive to DSB-inducing anticancer drugs [34]. This might result from HRR-mediated DSBR in pre-replicative DNA with no possibility to inhibit end resection by RIF1.
Grabarz et al. showed that the BLM (BLM RecQ like helicase) helicase suppressed long-range deletions generated by alternative end-joining [103]. BLM did so in an epistatic manner with TP53BP1 and RIF1. In turn, Zimmerman et al. showed that RIF1 impaired the resection factors CtIP (RB binding protein 8, endonuclease), BLM and EXO1 (exonuclease 1) and limited accumulation of BRCA1-BARD1 (BRCA1 associated RING domain 1) complexes at DSB sites. [104]. These results confirm the role of RIF1 as an important element of the DSBR control by TP53BP1 [97].
Bakr et al. showed that RIF1 acted epistatically with TP53BP1 to prevent DNA end resection at DSBs mediated by BRCA1/CtIP [105]. They also showed that RIF1 depletion in G1 cells caused accumulation of BRCA1, CtIP and RPA (replication protein A), but not RAD51 foci. Interestingly PARP1 (poly(ADP-ribose) polymerase 1)-dependent end joining (PARP1-EJ) was activated in RIF1-depleted G1 cells. Therefore, in G1 cells, in contrary to their S and G2 counterparts, the absence of RIF1 and TP53 is not sufficient to promote HRR and instead an error-prone PARP-EJ. In conclusion, Bart et al. proposed a mechanism in which RIF1 and TP53BP1 mediated the formation of a chromatin barrier around DSBs and promoted NHEJ. RIF1 was recruited along with, but independently of PTIP, and they could cooperate in end protection, but the exact mechanism of this cooperation is not known. As olaparib, a PARP1 inhibitor, is widely used in cancer therapy, RIF1 can be considered as a prognostic marker in olaparib-based anti-cancer treatment [106].
To search for the molecular details of RIF1 recruitment to DSB sites, Setiaputra et al. showed that RIF1 had a phosphopeptide-binding protein properties directly interacting with three TP53BP1 epitopes [107]. The RIF1-binding sites in TP53BP1 had two phosphorylated residues (S176 and S178) and their mutations abolished RIF1 accumulation into foci induced by ionizing radiation (IR), but the complete abrogation was observed only when an alternative mode of shieldin recruitment to DSB sites was also disabled. The authors concluded that RIF1 used phosphopeptide recognition to promote DSBR and modified shieldin action independently of TP53BP1 binding.
5.2. DNA End Resection
As mentioned, DNA end resection is a major effect of DSBR pathway choice. At the initial stage, MRE11 and CtIP are involved, but then it is accomplished by many proteins with a pronounced role of BRCA1 and cyclin-dependent kinase 1 CDK1 [108,109]. CDK1 phosphorylates CtIP at the G1→S transition to initiate end resection and direct DNA repair to HRR [108,110]. However, several other proteins may influence this process, first TP53BP1 that acts antagonistically to BRCA1, suppresses end resection and HRR and promotes NHEJ [111]. On the other hand, BRCA1 may dephosphorylate TP53BP1 to maintain HRR [112]. Therefore, TP53BP1 protects DNA ends against degradation at DSB sites. It does so with other proteins, including RIF1 and Pax transactivation domain-interacting protein (PTIP), which are recruited at DSB in a TP53BP1-dependent manner [113]. Therefore, RIF1 is loaded on the TP53BP1 scaffold to suppress NHEJ and direct DNA repair to HRR. RIF1 recruitment depends on TP53BP1 phosphorylation mediated by ATM [107]. This confirms multifaceted functionality of ATM as it is critical for the end resection process [114]. Exonuclease I is responsible for end resection in HRR and it may be recruited to DSB sites by the changes in chromatin structure promoted by the release of RIF1 from the TP53BP1-RIF1 complex and TP53BP1 repositioning [85].
Li et al. showed that DEAD box 1 (DDX1), an RNA helicase involved in DSBR, interacted and colocalized with RIF1 in interphase [115]. DDX1 was recruited to DSB sites in a RIF1-dependent manner and RIF1 depletion abolished DDX1-mediated HRR. The main conclusion of this study was that DDX1 might be responsible for the different consequences of RIF1 depletion has not completely the same consequence as TP53BP1 ablation in restoring HRR defects in BRCA1-deficient cells.
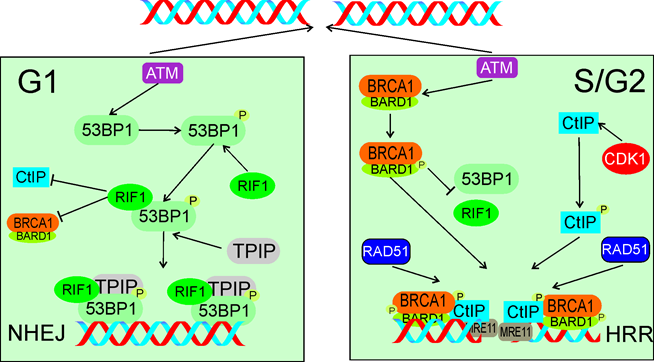
Figure 6. In response to DSBs in G1 (left panel) ATM phosphorylates TP53BP1 (53BP1), which associates with its critical effectors RIF1 and TPIP. These two proteins form a complex to block DNA end resection activity of CtIP and BRCA1/BARD1 and support NHEJ. In S/G2 (right panel) ATM phosphorylates BARD1 and CDK1 phosphorylates CtIP. Phosphorylated BRCA1/BARD1 prevents TP53BP1 binding by RIF1 and recruits phosphorylated CtIP and MRE11 to DSB sites to initiate end resection with subsequent recruitment of HRR machinery represented here by RAD51. Phosphorylation is marked by only single phosphate residue (P). The events presented in the right panel may also lead to microhomology-mediated end joining. All abbreviations are defined in the main text.
Escribano-Diaz et al. showed that TP53BP1 suppressed BRCA1 accumulation at DSB sites in G1 [116]. ATM-dependent phosphorylation of TP53BP1 recruited RIF1 and TPIP to DSB sites, which are critical effector of TP53BP1 in DSBR. These authors also showed that BRCA1 and CtIP antagonized RIF1 accumulation at DSB sites. PTIP contains the BRCT (BRCA1 C-terminal) domain, facilitating its interaction with TP53BP1 [117]. The relationship between TP53BP1, RIF1 and TPIP is not fully known, and likely complex [118]. Moreover, RIF1 depletion restored end resection and RAD51 (RAD51 recombinase), a critical HRR protein, loading in BRCA1-depleted cells. This work presented RIF1 and BRCA1 as critical elements of DSBR pathway choice to favor NHEJ in G1 phase and HRR in S phase of the cell cycle (Figure 6).
Although RIF1 promotes NHEJ by suppressing DNA end resection, there are several NHEJ variants with distinct mechanisms and proteins involved, so mechanisms of RIF1-mediated NHEJ promotion may be different for different NHEJ variants [119]. This issue was addressed by Han et al. who showed that the TP53BP1-RIF1-Artemis complex promoted alternative pathway of NHEJ (alt-NHEJ) by the retention of Artemis at DSB sites [120]. Earlier, Artemis was shown to be a downstream effector of the TP53BP1-PTIP pathway promoting processing DNA ends and the canonical NHEJ pathway [121]. These authors also observed that downregulation of RIF1 decreased alt-NHEJ in BRCA2-depleted cells to comparable level as in the case of TP53BP1 depletion. Isobe et al. reported that SCAI (suppressor of cancer cell invasion) bound TP53BP1 phosphorylated at S/TP sites and supported HRR [122]. SCAI gradually replaced RIF1 in the complex with TP53BP1 formed at DNA damage sites. Depletion of SCAI decreased accumulation of HRR factors, including BRCA1. Therefore, SCAI inhibits RIF1 to activate BRCA1-mediated repair, which may include alt-NHEJ and resection-dependent canonical NHEJ in G1, as well as HRR in S/G2.
5.3. Interaction with Shieldin
The protein complex shieldin was identified as a TP53BP1 effector that might be essential for TP53BP1-mediated suppression of end resection [123]. This complex consists of SHLD1 (shieldin complex subunit 1, C20orf196), SHLD2 (FAM35A, family with sequence similarity 35 member A), SHLD3 (CTC-534A2.2) and MAD2L2. Noodermeer et al. showed that shieldin localized at DSB sites in a TP53BP1- and RIF1-dependent manner and its SHLD2 subunit bound ssDNA protecting it from nucleolytic attack [124]. These authors also showed that SHELD3 directly interacted with RIF1, suggesting that this subunit recruited shieldin to TP53BP1-RIF1 bound to chromatin. These results were independently confirmed by Gao et al. [125]. Findlay et al. identified SHLD2 as an effector of MAD2L2 (mitotic arrest deficient 2 like 2, REV7) in NHEJ and showed that SHLD2-depeltion impaired NHEJ and compromised antibody diversification by class switching recombination in B cells [126]. MAD2L2 was identified as a factor controlling DNA repair at mammalian telomeres [127]. Further experiments showed that MAD2L2 displayed other DDR-related activities, including NHEJ promotion by inhibiting 5' end resection downstream of RIF1. SHLD2 accumulated at DSBs in dependence on TP53BP1, RIF1 and MAD2L2 and antagonized HRR by blocking DNA end resection. Therefore, SHLD2 may play an important role in the RIF1-mediated decisive mechanism of DSBR choice.
Isobe et al. showed that RIF1, but not shieldin, suppressed accumulation of CtIP at DSB sites immediately after damage [128]. These authors found that PP1, a known RIF1 effector in DNA replication, suppressed CtIP accumulation and limited the resection by the MRN complex. In conclusion, RIF1 inhibited end resection with PP1 before shieldin action to prevent HRR in early DDR.
Zhao et al. recently identified the RIF1 downstream effector complex ASTE1 (asteroid homolog 1) as a structure-specific DNA endonuclease, specifically resecting DNA 3’ overhangs, acting downstream of the shieldin complex [129]. Therefore, ASTE1 can be a new factor involved in the control of DSBR pathway choice by the TP53BP1-RIF1-shieldin signaling.
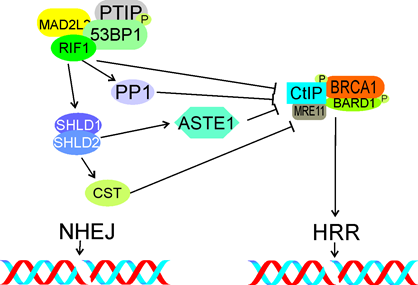
Figure 7. The TP53BP1 (53BP1)-RIF1-shieldin signaling in the choice of DSBR pathway in G1. RIF1 with phosphorylated TP53BP1 and assisted by MAD2L2 and TPIP, inhibits accumulation of phosphorylated CtIP and BRCA1 at DSB sites, preventing their interaction with MRE11, end resection and in consequence – inhibits HRR. Shieldin, represented here by the SDHLD1-SHLD2 complex, promotes NHEJ by interaction with NHEJ proteins (not shown). It may be also involved in CtIP/BRCA1 inhibition (not shown). The CST complex inhibits end resection and HRR downstream of shieldin, so does the ASTE complex. Phosphorylation is marked by only single phosphate residue (P). All abbreviations are defined in the main text.
Mirman et al. showed that the CST complex consisting of CTC1 (CST telomere replication complex component 1), STN1 (STN1 subunit of CST complex) and TEN1 (TEN1 subunit of CST complex), an accessory factor of the polymerase a-primase complex, was a downstream effector of the TP53BP1-RIF1-shieldin pathway regulating end resection [130]. Depletion of CST caused an increased end resection similarly to loss of RIF1, TP53BP1 or shieldin. Therefore, CST may assist RIF1, TP53BP1 and shieldin in DSBR regulation (Figure 7).
5.4. Epigenetics
Although changes in the epigenetic pattern associate with local changes in chromatin structure, we decided to present the involvement of RIF1 in these effects in separate sections for the sake of clarity.
DSBs trigger a series of ubiquitylation that contributes to the choice of DSBR pathway. This series includes the ubiquitylation of histone H2A by the RNF168 (ring finger protein 168) ligase and the subsequent recruitment of RIF1, which inhibits homologous recombination in G1 cells [131]. Partner and localizer of BRCA2 (BRCA2 DNA repair associated) (PALB2) forms oligomers with BRCA1/2 and this interaction must be tightly regulated to ensure suppression of HRR in G1 and its activation in S and early G2 [132,133]. Signaling pathway involved in the choice between HRR and NHEJ includes a join action of the E3 ubiquitin ligases RNF8 and RNF168 resulting in recruitment of the BRCA1-Abraxas-RAP80 (ubiquitin interaction motif containing 1)-MERIT40 (BRCA1-A, BRISC and BRCA1 A complex member 1) and TP53BP1 complexes [116]. TP53BP1 is recruited through its effectors RIF1 and MAD2L2 and inhibits end resection.
As HRR requires the presence of an intact template of replicated DNA, cells have evolved mechanisms to block HRR in G1, when after mitosis, a homology partner may be difficult to find (reviewed in [134]). When cells are about to enter S phase, these blocking mechanisms are freed to activate HRR. However, HRR in non-replicated DNA in S phase may promote aberrant DNA structures that may lead to stalling or collapsing of replication fork. Therefore, the choice between NHEJ and HRR may be influenced by the replication status of a DNA fragment – non-replicated vs. replicated. In S phase NHEJ can process DSBs in pre-replicative DNA and HRR – in post-replicative DNA.
Simonetta et al. showed that MAD2L2 (mitotic arrest deficient 2 like 2, REV7) was recruited to DSB sites in H4K20 dimethylated chromatin by forming a complex with TP53BP1 and RIF1 [135]. This complex inhibited the accumulation of BRCA1 at these sites. Simonetta et al. also showed that the choice of DSBR was determined by the replication status of a DNA fragment and epigenetic mechanisms. In non-replicated DNA, the TP53BP1-RIF1-MAD2L2 complex was formed at DSB sites with a subsequent exclusion of BRCA1 at di-methylated lysine 20 of histone 4 (H4K20me2), designating NHEJ as a preferred DSBR pathway. When this DNA fragment was replicated, it caused the dilution of H4K20me2 resulting in TP53BP1-RIF1-MAD2L2 releasing, access of BRCA1, end resection and HRR. Therefore, the H4K20 methylation status in pre-replicative and post-replicative DNA may be an intrinsic mechanism of DSBR pathway choice through the binding/releasing of the TP53BP1-RIF1-MAD2L2 complex.
Drane et al. observed that RIF1 was necessary to separate TP53BP1 from Tudor interacting repair regulator (TIRR) [98]. Interaction of TIRR with TP53BP1 shielded its H4K20me2-binding motif, necessary for TP53BP1 recruitment to DSB sites. This work reveals further details of TP53BP1-RIF1 interaction in DSBR, suggesting that epigenetic pattern of chromatin may play a role. Therefore, local chromatin changes may play a role in the RIF1 signaling in DSBR.
Kumar and Cheok showed that in response to DNA damage RIF1 was SUMOylated [136]. This process was primarily mediated by protein inhibitor of activated STAT (PIAS4), a SUMO E3 ligase. Furthermore, PIAS4 downregulation caused impaired RIF1 SUMOylation, defects in the disassembly of DDR-related RIF1 foci, and abolished UHRF1 (ubiquitin like with PHD and ring finger domains 1)-dependent ubiquitination of RIF1, impairing turnover of the RIF1 protein. Kumar and Cheok underlined the role of SUMOylation in disassembly of the RIF1 DDR-related foci showing that disturbances in this process might lead to DSB induction and concluded that PIAS4 promoted genomic stability by temporal regulation of removal of RIF1 from DSB sites.
5.5. Local 3D Chromatin Changes
CSB (ERCC excision repair 6, chromatin remodeling factor) is involved in DSBR and this involvement is mediated by the interaction with RIF1 through its winged helix domain in S phase [137]. At DSB sites, CBS displaces histones, limiting RIF1 and its effector MAD2L2, but supporting BRCA1 accumulation. As regulation of the DSBR pathway choice by RIF1 is mediated by changes in chromatin, proteins of chromatin remodeling may modulate this regulation, especially when they directly interact with RIF1.
These studies provoke several questions about local changes in 3D chromatin in the vicinity of a DSB, their specificity to replicating DNA and the involvement of RIF1. These questions have been addressed by “classical” microscopy, but none of them has been fully answered [138,139]. Development of 3D structured illumination microscopy (3D-SIM) and higher-resolution imaging by stimulation depletion (STED) microscopy provided adequate tools to address these problems [140,141]. Sites of DSBs are organized within chromatin in nanodomains containing elementary DDR-related protein foci (nano-foci) [142]. Ochs et al. used live-cell STED microscopy and 3D-SIM and identified 4-7 subdomains in a typical TP53BP1 focus [143]. These subdomains were divided into TP53BP1 nano- and microdomains (TP53BP1-NDs and TP53BP1-MD, respectively). RIF1 depletion caused breakage of TP53BP1-MDs into disordered and elongated structures featured by misaligned TP53BP1-NDs. These data suggest that RIF1 forms an autonomous unit with TP53BP1 to stabilize TP53BP1-NDs into an ordered chromatin architecture. At the DSB sites, RIF1 localized to the chromatin between adjacent TP53BP1-NDs and its depletion caused inability of TP53BP1-NDs to organize into higher order structures, TP53BP1-MDs. Importantly, disruption of the shieldin complex did not interfere with spatial organization of TP53BP1-NDs. Therefore, RIF1 recruited to DSBs may play a specific role in the stabilization of chromatin mediated by the creation of TP53BP1-NDs. These studies also confirmed that RIF1 cooperated with cohesin to maintain chromatin structure at DSB sites and that RIF1 depletion favored DNA end resection at these sites, as evidenced by BRCA1 expansion. RIF1 was not sufficient to change the chromatin structure, but it was primary responsible for the stabilization of the structure around a DSB. Finally, the authors speculated that the association of RIF1 with TP53BP1 might have evolved to preserve epigenetic markers hidden in 3D chromatin structure challenged by DSBs.
- RIF1 in Reactivation of Impaired Replication Fork
Activation of the intra-S checkpoint blocks the cell cycle and activates HRR to restart arrested replication fork (reviewed in [144,145]).
Stalled or collapsed replication fork disturbs RT and its reactivation is essential for the cell fate, as apart from impaired RT, damaged fork may have serious delayed consequence, including genomic instability contributing to several pathologies [79]. Therefore, cells evolved pathways to restart damaged replication fork, which can be broadly divided into occurring with or without DSB intermediates [146,147].
An important example illustrating the interconnection between DNA replication and DSBR is the case of replication fork encountering interstrand cross-links (ICLs) (reviewed in [148]). ICLs are induced by an agent covalently bound to the two strands of a DNA molecule, impairing strands separation, a prerequisite in DNA replication, transcription and recombination. There is not a uniform DNA repair pathway targeting ICLs. The Fanconi anemia network, nucleotide excision repair, HRR and translesion synthesis (TLS) are involved in ICLs repair with DSBs as an intermediate (reviewed in [149]). Even an accurate repair of ICLs may interfere with RT if the cell is not given enough time for such repair.
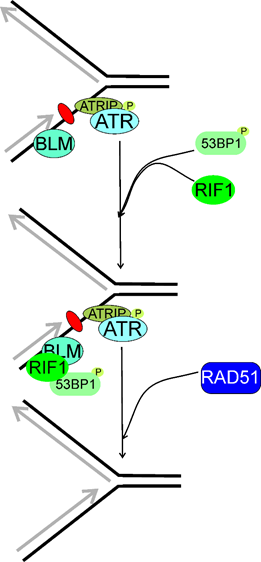
Figure 8. Replication timing regulatory factor 1 (RIF1) may be involved in the reactivation of stalled replication fork, which is recognized by ATR (ATM-and-RAD3-related) acting in concert with its partner, ATRIP (ATR interacting protein). RIF1 associates with phosphorylated (P) T53BP1 (53BP1, tumor protein p53 binding protein) and colocalizes with the BLM RecQ helicase foci. Then, the homologous recombination repair machinery symbolized here by RAD51 is recruited to recover the stalled fork. Here, stalling of replication fork results from a single DNA lesion (purple oval) in the lagging strand, but many other DNA damages may induce such effects. Moreover, RIF1 may stabilize replication fork and prevent its collapse or reversal, but the exact mechanism of this RIF1 activity is not completely known. T53BP1 might not be of the RIF1-BLM complex and might be separately recruited to stalled replication fork.
Xu et al. showed that RIF1 was a part of a multiprotein complex to maintain genomic stability, the BLM complex, containing the BLM helicase [21,150]. Human RIF1 and BLM were recruited to ICLs with the same rate and majority of them colocalized [21] (Figure 8). RIF1 cooperated with BLM to recover stalled replication fork. The stability of RIF1 depended on the BLM complex. Although a direct interaction between the C-terminal domain of RIF1 with the BLM complex, RIF1 was also recruited, while less efficiently, to stalled replication fork in the absence of BLM. Although previously Buonomo et al. showed that RIF1 colocalized with TP53BP1 and a fraction of BLM, Xu et al observed that TP53BP1 was not a part of the BLM complex and might be separately recruited to the stalled replication fork [32,151]. In conclusion, RIF1 and BLM worked in the same pathway to prevent damage accumulation and promoted reactivation of stalled replication forks.
The presence of two distinct fork recovery pathways controlled by TP53BP1 and BRCA1 independently of DSBR was shown [151]. Xu et al. showed that TP53BP1 and RIF1 played a NHEJ-independent role in response to replication stress. The absence of TP53BP1-induced hypersensitivity to replication stress that was suppressed by the BRCA1 deletion. TP53BP1 promoted the fast kinetics of fork restart, whereas BRCA1 was involved in slow fork reactivation. The authors claimed that TP53BP1 was dispensable to DSBR at broken replication fork, but BRCA1 promoted MUS81 (MUS81 structure-specific endonuclease subunit)-coupled break-induced recombination (BIR), a subpathway of HRR.
The importance of DSBs and their repair for RT provokes the question about timing of DSBR, especially in meiosis [152]. Moreover, resuming of DNA replication in the case of ICLs repair and some other circumstances is linked with DSBs affecting RT with a strong causal association with genomic instability and cancer (reviewed in [81]).
Mouse RIF1 deficiency induced an increased aphidicolin-evoked replication stress, a defect in the intra-S checkpoint, and an accumulation of DNA damage in S phase [33]. RIF1 localized at sites with stalled replication fork, mostly at pericentromeric chromatin. A decrease in HRR efficacy was observed in cells with reduced RIF1 levels. This was associated with aggregation of aberrant RAD51. It was shown that on ATR activation and TP53BP1 recruitment, RIF1 localized at a subset of stalled replication forks preferentially in mid-late S phase, and regulated HRR to resume replication. RIF1 deletion induced a chromatid-type genome instability. It was hypothesized that RIF1 regulated RAD51-dependent HRR in the regions of the genome that are difficult to replicate, including repeated DNA sequences, and consequently prone to replication stress. RIF1 foci at stalled forks colocalized with the BLM RecQ helicase, but not all BLM foci overlapped those of RIF1.
Xu et al. showed that TP53BP1-RIF1 and BRCA1 played different than DSBR roles in the reactivation of replication fork [151] They observed that only the defect in fork restart but not DSB repair in the TP53BP1-dpleted cells was ameliorated by the interruption of BRCA1. The antagonistic roles of TP53BP1 and BRCA1 in replication restart are somehow similar to their roles in DSBR, in which both proteins counteract functions in DSBR. Furthermore, end resection and fork cleavage are the decision steps of DSBR pathway choice and fork restart pathway, respectively. The former is initiated by the CtIP-MRN endonuclease complex, the latter is performed by the MUS-SLX (structure-specific endonuclease subunit) endonuclease complex. The authors hypothesized that TP53BP1 and RIF1 might display a common mechanism in blocking DSBs resection and preventing fork cleavage. This mechanism might include the formation of a higher-order chromatin structure to limit the access of BRCA1-recruited nucleases [98]. This function is apparently in conflict with the ability of RIF1 to recruit the downstream proteins, such as BLM. BRCA1 might disturb the chromatin structures needed for TP53BP1-RIF1 accumulation as BRCA1 displays a chromatin-decondensation activity [153]
Matoo et al. showed that myeloid cell leukemia 1 (MCL-1) is essential for the survival of cells exposed to ionizing radiation (IR) [154]. Depletion of MCL-1 in IR-exposed hematopoietic stem cells increased genomic instability and the number of residual TP53BP1 and RIF1 foci and decreased formation of foci related to HRR. Analogous effect was observed in MCL-1-depleted cells with stalled replication fork induced by hydroxyurea. In conclusion, MCL-1 depletion increases TP53BP1 and RIF1 colocalization and inhibits BRCA1 recruitment and HRR needed for repairing DSBs caused by IR or resolution of stalled replication fork.
To look for a mechanism linking the involvement of RIF1 in RT and DSBR, Saito et al. exposed HeLa cells to high doses of IR [155]. They observed that IR suppressed HRR and this effect was mediated by RIF1. IR did not affect NHEJ. At low IR doses BRCA1 inhibited RIF1 in S/G2 phase. At high IR doses RIF1 dephophorylated the MCM helicase resulting in suppression of initiation of DNA replication. Dephosphorylation of MCM results in inhibition of both end resection and HRR even independently of RIF1 [156]. The authors concluded that MCM might be a possible link between DNA replication and HRR and RIF1 controlled suppression of both initiation of replication and HRR at high IR doses.
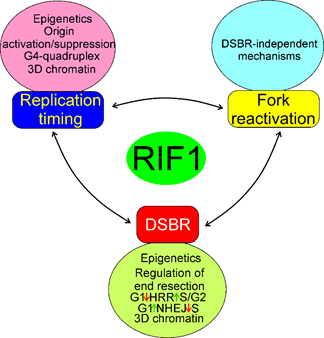
Figure 9. Mechanisms of the involvement of RIF1 in the regulation of RT, fork reactivation and DSBR. Epigenetics here represents changes in the cellular epigenetic pattern associated with RIF1 action and they, along with concomitant changes in chromatin structure, are essential for all processes of RIF1. All three effects: RT, fork reactivation and DSBR are mutually dependent, but the extent and details of this dependency are to be established. All abbreviations are defined in the main text.
In summary, RIF may be involved in RT, DSBR and fork reactivation by mechanisms that may partly overlap, but the exact determination of the extent of that overlapping requires further research. In general, RIF1 may regulate RT by changes in the 3D structure of chromatin, binding of G4-quadruplexes, common in telomeres, and activation deactivation of proteins involved in the initiation of DNA replication, including the MCM helicase (Figure 9). Impaired functions of replication fork, resulting from its stalling, slowing or collapsing strongly affect RT and RIF1 may reactivate the impaired fork through mechanisms that can be either related to or independent of its functions in DSBR. RIF1 may play an important role in the choice of DSBR pathway through its association with phosphorylated TP53BP1 and suppression of DNA end resection in G1 phase of the cell cycle, required in HRR, but not needed in NHEJ. On the other hand, lack of such action in S/G2 phase of the cell cycle promotes HRR.
7. Conclusion and Perspectives
Replication timing is an essential program to ensure timing, complete and accurate replication of the human genome and adjust DNA replication to other ongoing cellular processes. RIF1 is a master regulator of RT in human cells, but it is a multifaceted protein whose some essential functions may be unknown.
Apart from RT regulation, RIF1 is involved in DSBR and reactivation of damaged replication fork. The involvement of RIF1 in DSBR is mainly associated with the role of RIF1 in the repair choice, but some studies suggest that it can be also involved in the executional phase of DNA repair. There is no doubt, that these functions may overlap, but the extent of overlapping and shared mechanisms of these RIF1 activities are poorly known. Therefore, future research addressing futher details of common regulation of RT, DSBR and reactivation of damaged replication fork are needed. Moreover, as DNA end resection is a key element in SSA, a third main DSBR pathway, and the main mechanism underlying DSBR pathway choice by RIF1, the involvement of RIF1 in SSA regulation is justified to study.
The data obtained so far suggest that the structure of chromatin may be essential for all known aspects of RIF1 activity. It is not surprising as changes in 3D structure of chromatin are inexcitrably associated with RT, DSBR and fork reactivation. As shown, the development of new imaging methods, including high-resolution microscopy, has a high potential to reveal new details of the involvement of RIF1 in 3D chromatin changes associted with its activites.
As yeast Rif1 phosphorylation sites were shown to play an important role in vital yeast function, it is justified to explore the role of corresponding sites of human RIF1 [24].
When impaired, all three phenomena: RT, DSBR and fork reactivation may contribute to genomic instability, which is typical for most, if not all, cancer cells. Therefore, RIF1 have a potential in cancer diagnosis and therapy [35-37]. Surely, the immediate question in this regard is whether RIF1 plays a role of oncogene or tumor suppressor gene and the answer is almost equally immediate as RIF1 is an effector of TP53BP1, which, in turn, positively regulates TP53, a tumor suppressor [157,158]. However, the role of TP53-TP53BP1 interaction in cancer transformation is not completely clear, so the corresponding role of RIF1 should be further explored [159].
When cell encounters a lesion slowing, stalling or colapsing replication fork, it may decide to baypass that lesion, which may cause mutations contributing to pathological consequences [144]. A potential role of RIF1 in this process has not been explored yet.
Distinc DSBR functions of RIF1 in replicated and non-replicated DNA are another key element to better understand its diverse actions.
In summary, RIF1 display several activities, which are mainly associated with its involvement in RT, DSBR and reactivation of damaged replication fork. This involvement is underlined by the interaction with many other proteins, including initiators of DNA replication, cell cycle regulators, DDR proteins, modifiers of epigenetic pattern and chromatin organization. This makes RIF1 as an important element in physiology and pathology with a potential in the therapy of many diseases.
References
- Dimitrova, D.S.; Gilbert, D.M. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell 1999, 4, 983–993.
- Vouzas, A.E.; Gilbert, D.M. Mammalian DNA replication timing. Cold Spring Harb. Perspect. Biol. 2021, 13, a040162.
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107.
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA double-strand break repair: An ever-growing toolbox. Front. Mol. Biosci. 2020, 7, 24.
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714.
- Fontana, G.A.; Rass, U. Compartmentalized DNA repair: Rif1 S-acylation links DNA double-strand break repair to the nuclear membrane. Mol. Cell. Oncol. 2019, 6, e1648025.
- Gnan, S.; Flyamer, I.M.; Klein, K.N.; Castelli, E.; Rapp, A.; Maiser, A.; Chen, N.; Weber, P.; Enervald, E.; Cardoso, M.C.; et al. Nuclear organisation and replication timing are coupled through RIF1-PP1 interaction. Nat. Commun. 2021, 12, 2910.
- Nathanailidou, P.; Taraviras, S.; Lygerou, Z. Chromatin and nuclear architecture: Shaping DNA replication in 3D. Trends Genet. 2020, 36, 967–980.
- Smogorzewska, A.; de Lange, T. Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 2004, 73, 177–208.
- Toteva, T.; Mason, B.; Kanoh, Y.; Brøgger, P.; Green, D.; Verhein-Hansen, J.; Masai, H.; Thon, G. Establishment of expression-state boundaries by Rif1 and Taz1 in fission yeast. Proc. Natl. Acad. Sci. USA 2017, 114, 1093–1098.
- Zhou, Z.; Wang, L.; Ge, F.; Gong, P.; Wang, H.; Wang, F.; Chen, L.; Liu, L. Pold3 is required for genomic stability and telomere integrity in embryonic stem cells and meiosis. Nucleic Acids Res. 2018, 46, 3468–3486.
- Daley, J.M.; Sung, P. RIF1 in DNA break repair pathway choice. Mol. Cell 2013, 49, 840–841.
- Hayano, M.; Kanoh, Y.; Matsumoto, S.; Renard-Guillet, C.; Shirahige, K.; Masai, H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 2012, 26, 137–150.
- Kanoh, Y.; Matsumoto, S.; Fukatsu, R.; Kakusho, N.; Kono, N.; Renard-Guillet, C.; Masuda, K.; Iida, K.; Nagasawa, K.; Shirahige, K.; et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015, 22, 889–897.
- Kumar, R.; Cheok, C.F. RIF1: A novel regulatory factor for DNA replication and DNA damage response signaling. DNA Repair 2014, 15, 54–59.
- Mattarocci, S.; Hafner, L.; Lezaja, A.; Shyian, M.; Shore, D. Rif1: A conserved regulator of DNA replication and repair hijacked by telomeres in yeasts. Front. Genet. 2016, 7, 45.
- Hardy, C.F.; Sussel, L.; Shore, D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992, 6, 801–814.
- Silverman, J.; Takai, H.; Buonomo, S.B.; Eisenhaber, F.; de Lange, T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 2004, 18, 2108–2119.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078.
- Xu, L.; Blackburn, E.H. Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J. Cell Biol. 2004, 167, 819–830.
- Xu, D.; Muniandy, P.; Leo, E.; Yin, J.; Thangavel, S.; Shen, X.; Ii, M.; Agama, K.; Guo, R.; Fox, D., 3rd; et al. Rif1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. EMBO J. 2010, 29, 3140–3155.
- Kobayashi, S.; Fukatsu, R.; Kanoh, Y.; Kakusho, N.; Matsumoto, S.; Chaen, S.; Masai, H. Both a unique motif at the C terminus and an N-terminal heat repeat contribute to G-quadruplex binding and origin regulation by the Rif1 protein. Mol. Cell. Biol. 2019, 39, e00364-18.
- Moriyama, K.; Lai, M.S.; Masai, H. Interaction of Rif1 protein with G-quadruplex in control of chromosome transactions. Adv. Exp. Med. Biol. 2017, 1042, 287–310.
- Wang, J.; Zhang, H.; Al Shibar, M.; Willard, B.; Ray, A.; Runge, K.W. Rif1 phosphorylation site analysis in telomere length regulation and the response to damaged telomeres. DNA Repair 2018, 65, 26–33.
- Sukackaite, R.; Jensen, M.R.; Mas, P.J.; Blackledge, M.; Buonomo, S.B.; Hart, D.J. Structural and biophysical characterization of murine rif1 C terminus reveals high specificity for DNA cruciform structures. J. Biol. Chem. 2014, 289, 13903–13911.
- Sreesankar, E.; Senthilkumar, R.; Bharathi, V.; Mishra, R.K.; Mishra, K. Functional diversification of yeast telomere associated protein, Rif1, in higher eukaryotes. BMC Genom. 2012, 13, 255.
- Masai, H.; Fukatsu, R.; Kakusho, N.; Kanoh, Y.; Moriyama, K.; Ma, Y.; Iida, K.; Nagasawa, K. Rif1 promotes association of G-quadruplex (G4) by its specific G4 binding and oligomerization activities. Sci. Rep. 2019, 9, 8618.
- Masai, H.; Kanoh, Y.; Moriyama, K.; Yamazaki, S.; Yoshizawa, N.; Matsumoto, S. Telomere-binding factors in the regulation of DNA replication. Genes Genet. Syst. 2018, 92, 119–125.
- Alavi, S.; Ghadiri, H.; Dabirmanesh, B.; Moriyama, K.; Khajeh, K.; Masai, H. G-quadruplex binding protein Rif1, a key regulator of replication timing. J. Biochem. 2021, 169, 1–14.
- Garzón, J.; Ursich, S.; Lopes, M.; Hiraga, S.I.; Donaldson, A.D. Human RIF1-protein phosphatase 1 prevents degradation and breakage of nascent DNA on replication stalling. Cell Rep. 2019, 27, 2558–2566.e2554.
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; de Boer, H.R.; Demmers, J.; van Vugt, M.; Ray Chaudhuri, A. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287.
- Li, P.; Wang, L.; Bennett, B.D.; Wang, J.; Li, J.; Qin, Y.; Takaku, M.; Wade, P.A.; Wong, J.; Hu, G. Rif1 promotes a repressive chromatin state to safeguard against endogenous retrovirus activation. Nucleic Acids Res. 2017, 45, 12723–12738.
- Buonomo, S.B.; Wu, Y.; Ferguson, D.; de Lange, T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J. Cell Biol. 2009, 187, 385–398.
- Wang, H.; Zhao, A.; Chen, L.; Zhong, X.; Liao, J.; Gao, M.; Cai, M.; Lee, D.H.; Li, J.; Chowdhury, D.; et al. Human RIF1 encodes an anti-apoptotic factor required for DNA repair. Carcinogenesis 2009, 30, 1314–1319.
- Liu, Y.B.; Mei, Y.; Long, J.; Zhang, Y.; Hu, D.L.; Zhou, H.H. RIF1 promotes human epithelial ovarian cancer growth and progression via activating human telomerase reverse transcriptase expression. J. Exp. Clin. Cancer Res. 2018, 37, 182.
- Mei, Y.; Liu, Y.B.; Cao, S.; Tian, Z.W.; Zhou, H.H. RIF1 promotes tumor growth and cancer stem cell-like traits in NSCLC by protein phosphatase 1-mediated activation of Wnt/β-catenin signaling. Cell Death Dis. 2018, 9, 942.
- Mei, Y.; Liu, Y.B.; Cao, S.; Tian, Z.W.; Zhou, H.H. Correction: RIF1 promotes tumor growth and cancer stem cell-like traits in NSCLC by protein phosphatase 1-mediated activation of Wnt/β-catenin signaling. Cell Death Dis. 2021, 12, 812.
- Howarth, K.D.; Blood, K.A.; Ng, B.L.; Beavis, J.C.; Chua, Y.; Cooke, S.L.; Raby, S.; Ichimura, K.; Collins, V.P.; Carter, N.P.; et al. Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene 2008, 27, 3345–3359.
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274.
- Fu, H.; Baris, A.; Aladjem, M.I. Replication timing and nuclear structure. Curr. Opin. Cell Biol. 2018, 52, 43–50.
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 721–737.
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374.
- Chong, J.P.; Mahbubani, H.M.; Khoo, C.Y.; Blow, J.J. Purification of an MCM-containing complex as a component of the DNA replication licensing system. Nature 1995, 375, 418–421.
- Boos, D.; Ferreira, P. Origin firing regulations to control genome replication timing. Genes 2019, 10, 199.
- MacNeill, S.A. Structure and function of the GINS complex, a key component of the eukaryotic replisome. Biochem. J. 2010, 425, 489–500.
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961.
- Friedman, K.L.; Diller, J.D.; Ferguson, B.M.; Nyland, S.V.; Brewer, B.J.; Fangman, W.L. Multiple determinants controlling activation of yeast replication origins late in S phase. Genes Dev. 1996, 10, 1595–1607.
- Ding, Q.; Koren, A. Positive and negative regulation of DNA replication initiation. Trends Genet. 2020, 36, 868–879.
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat. Commun. 2018, 9, 3704.
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132.
- Gilbert, D.M. Cell fate transitions and the replication timing decision point. J. Cell Biol. 2010, 191, 899–903.
- Cvetic, C.; Walter, J.C. Eukaryotic origins of DNA replication: Could you please be more specific? Semin. Cell Dev. Biol. 2005, 16, 343–353.
- Raghuraman, M.K.; Winzeler, E.A.; Collingwood, D.; Hunt, S.; Wodicka, L.; Conway, A.; Lockhart, D.J.; Davis, R.W.; Brewer, B.J.; Fangman, W.L. Replication dynamics of the yeast genome. Science 2001, 294, 115–121.
- Farago, M.; Rosenbluh, C.; Tevlin, M.; Fraenkel, S.; Schlesinger, S.; Masika, H.; Gouzman, M.; Teng, G.; Schatz, D.; Rais, Y.; et al. Clonal allelic predetermination of immunoglobulin-κ rearrangement. Nature 2012, 490, 561–565.
- Mostoslavsky, R.; Singh, N.; Tenzen, T.; Goldmit, M.; Gabay, C.; Elizur, S.; Qi, P.; Reubinoff, B.E.; Chess, A.; Cedar, H.; et al. Asynchronous replication and allelic exclusion in the immune system. Nature 2001, 414, 221–225.
- Chess, A. Random and non-random monoallelic expression. Neuropsychopharmacology 2013, 38, 55–61.
- Chess, A.; Simon, I.; Cedar, H.; Axel, R. Allelic inactivation regulates olfactory receptor gene expression. Cell 1994, 78, 823–834.
- Blumenfeld, B.; Masika, H.; Farago, M.; Yehuda, Y.; Halaseh, L.; Vardi, O.; Rapoport, R.; Levin-Klein, R.; Cedar, H.; Bergman, Y.; et al. Chromosomal coordination and differential structure of asynchronous replicating regions. Nat. Commun. 2021, 12, 1035.
- Hadjadj, D.; Denecker, T.; Maric, C.; Fauchereau, F.; Baldacci, G.; Cadoret, J.C. Characterization of the replication timing program of 6 human model cell lines. Genom. Data 2016, 9, 113–117.
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.W.; Lyou, Y.; Townes, T.M.; Schübeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245.
- Donley, N.; Thayer, M.J. DNA replication timing, genome stability and cancer: Late and/or delayed DNA replication timing is associated with increased genomic instability. Semin. Cancer Biol. 2013, 23, 80–89.
- Zhang, H.; Petrie, M.V.; He, Y.; Peace, J.M.; Chiolo, I.E.; Aparicio, O.M. Dynamic relocalization of replication origins by Fkh1 requires execution of DDK function and Cdc45 loading at origins. eLife 2019, 8, e45512.
- Tanaka, S.; Nakato, R.; Katou, Y.; Shirahige, K.; Araki, H. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr. Biol. 2011, 21, 2055–2063.
- Briu, L.M.; Maric, C.; Cadoret, J.C. Replication stress, genomic instability, and replication timing: A complex relationship. Int. J. Mol. Sci. 2021, 22, 4764.
- Alver, R.C.; Chadha, G.S.; Gillespie, P.J.; Blow, J.J. Reversal of DDK-mediated MCM phosphorylation by Rif1-PP1 regulates replication initiation and replisome stability independently of ATR/Chk1. Cell Rep. 2017, 18, 2508–2520.
- Pope, B.D.; Gilbert, D.M. The replication domain model: Regulating replicon firing in the context of large-scale chromosome architecture. J. Mol. Biol. 2013, 425, 4690–4695.
- Klein, K.N.; Zhao, P.A.; Lyu, X.; Sasaki, T.; Bartlett, D.A.; Singh, A.M.; Tasan, I.; Zhang, M.; Watts, L.P.; Hiraga, S.I.; et al. Replication timing maintains the global epigenetic state in human cells. Science 2021, 372, 371–378.
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770.
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380.
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 2017, 169, 930–944.e922.
- Oldach, P.; Nieduszynski, C.A. Cohesin-mediated genome architecture does not define DNA replication timing domains. Genes 2019, 10, 196.
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56.
- Miura, H.; Takahashi, S.; Shibata, T.; Nagao, K.; Obuse, C.; Okumura, K.; Ogata, M.; Hiratani, I.; Takebayashi, S.I. Mapping replication timing domains genome wide in single mammalian cells with single-cell DNA replication sequencing. Nat. Protoc. 2020, 15, 4058–4100.
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol. Cell 2016, 61, 260–273.
- Yamazaki, S.; Hayano, M.; Masai, H. Replication timing regulation of eukaryotic replicons: Rif1 as a global regulator of replication timing. Trends Genet. 2013, 29, 449–460.
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637.
- Hou, Y.; Li, F.; Zhang, R.; Li, S.; Liu, H.; Qin, Z.S.; Sun, X. Integrative characterization of G-Quadruplexes in the three-dimensional chromatin structure. Epigenetics 2019, 14, 894–911.
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic instability in cancer: Teetering on the limit of tolerance. Cancer Res. 2017, 77, 2179–2185.
- Keszthelyi, A.; Minchell, N.E.; Baxter, J. The causes and consequences of topological Stress during DNA replication. Genes 2016, 7, 134.
- Yoshida, K.; Fujita, M. DNA damage responses that enhance resilience to replication stress. Cell. Mol. Life Sci. 2021, 1–11.
- Ui, A.; Chiba, N.; Yasui, A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020, 111, 1443–1451.

