Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Victoria Smith Arnesen | + 3982 word(s) | 3982 | 2021-10-15 09:44:37 | | | |
| 2 | Martha Chekenya | Meta information modification | 3982 | 2021-10-27 15:43:02 | | | | |
| 3 | Conner Chen | Meta information modification | 3982 | 2021-10-28 03:35:04 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Arnesen, V.S.; Chekenya, M. Designer T and NK Cells in Glioblastoma Immunotherapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/15471 (accessed on 28 July 2026).
Arnesen VS, Chekenya M. Designer T and NK Cells in Glioblastoma Immunotherapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/15471. Accessed July 28, 2026.
Arnesen, Victoria Smith, Martha Chekenya. "Designer T and NK Cells in Glioblastoma Immunotherapy" Encyclopedia, https://encyclopedia.pub/entry/15471 (accessed July 28, 2026).
Arnesen, V.S., & Chekenya, M. (2021, October 27). Designer T and NK Cells in Glioblastoma Immunotherapy. In Encyclopedia. https://encyclopedia.pub/entry/15471
Arnesen, Victoria Smith and Martha Chekenya. "Designer T and NK Cells in Glioblastoma Immunotherapy." Encyclopedia. Web. 27 October, 2021.
Copy Citation
Glioblastoma (GBM) is the most prevalent, aggressive primary brain tumour with a dismal prognosis. Gene editing technologies are a game changer, enabling design of novel molecular-immunological treatments to be used in combination with chemoradiation, to achieve long lasting survival benefits for patients. Designer T and NK cells are a modality within immunotherapy that manipulates receptor-ligand interactions to enhance cells of the immune system to destroy cancer more effectively. Patient’s own immune cells are isolated, genetically modified to improve responses against cancer cells, expanded, and subsequently reintroduced into the individual.
glioblastoma
genomic heterogeneity
natural killer cells
T cells
chimeric antigen receptor
CRISPR/Cas9
immunotherapy
1. T Cell Patrols with a License to Kill
Patrolling leukocytes detect mutated, early transformed cells, deem them sufficiently “non-self” and co-ordinately eliminate them, as predicted by the cancer immunosurveillance concept [1]. As cells undergo oncogenesis, neoantigens are released and captured on major histocompatibility complex (MHC)/ human leukocyte antigen (HLA) of dendritic cells (DCs) that subsequently mature and migrate to central lymphoid organs. Here, the peptide neoantigen on the DCs’ MHC is presented to the awaiting CD4+ or CD8+ T cell receptor (TCR) complex. Subsequently, binding of the CD28 co-stimulatory receptor to the DCs’ CD80/86 receptor fully activates the cytotoxic T cells which then migrate to infiltrate the tumour and kill the cells by locally releasing perforin and granzymes [2][3]. These lymphocytes successfully eliminate the genetically unstable tumour cells with intrinsically high immunogenicity [4] through a series of successive stages [5]. The T cells also effectively terminate their activation and proliferation as a means of avoiding autoimmunity, resulting in different phenotypes that either further activate Th1 immune responses or suppress via Th2-driven responses. Surface receptors such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1) and nuclear transcription factors attenuate T cell responses, where CTLA-4 competes with CD28 for binding to CD80/86, resulting in inhibitory downstream signalling [6]. PD-1 is an immunoinhibitory receptor that stymies lymphocyte proliferation and cytokine secretion when bound to its membrane-bound or secreted ligands, PD-L1 or PD-L2, expressed by both immune and tumour cells [7]. In addition, activated T cells can express an inducible co-stimulator (iCOS), a surface receptor that is structurally and functionally similar to CD28 and enhances expression of Th2-related interleukin (IL)-10 rather than immune activating IL-2 [8]. This immunosuppressive helper T cell phenotype can be further induced by the zinc-finger transcription factor GATA3, which regulates Th2 cytokine expression [9]. Infiltrating CD8+ T cells in GBM have been shown to bear an exhausted phenotype, with upregulated expression of PD-1, LAG-3, TIGIT and CD39 [10][11][12].
GBM is considered an immunologically “cold tumour” due to the immunosuppressive microenvironment [12][13] and few immunogenic tumour-associated antigens (TAAs) [14]. The rapidly growing tumour alters the balance of interaction between cancer and immune cells, by outstripping its metabolic resources and shifting to glycolysis [15]. Cancer cells also recruit and alter nearby stromal cells to aid the tumour cells in avoiding immune detection and destruction [16][17]. Competition for glucose results in low pH, which creates a hostile tumour microenvironment [18][19][20] and drives a local increase in immunosuppressive stromal cells (Figure 1). These cells secrete immune inhibitory growth factors and cytokines, including vascular endothelial growth factor (VEGF), which is primarily produced by microglia, myeloid-derived suppressor cells (MDSCs) and tumour-associated macrophages (TAMs). TAMs and regulatory T cells such as the CD8+ CD28- FoxP3+ Treg cells also secrete immunosuppressive IL-10 [12] which downregulates interferon gamma (IFNγ) and MHC I through the JAK/STAT pathway [21]. Accumulating MDSCs are rendered in an immature state [22], secreting IL-13, which when bound to IL13Rα2, send anti-inflammatory signals to promote the alternative M2 phenotype in macrophages, causing them to lose their cytotoxic function and instead become immunosuppressive. Tumour cells aid in this immunosuppression by secreting transforming growth factor β (TGFβ) and expressing both membrane-bound and soluble PD-L1, inducing exhaustion in T and NK cells [23][24]. Moreover, the molecular heterogeneity of GBM, observed as cells with wildly dissimilar molecular genetic aberrations within the same tumour mass [25][26] allows the tumour microenvironment to evolve and adapt to the immunological selection pressure. These conditions lead to immune escape and an immune-edited microenvironment that is refractory to killing by cytotoxic lymphocytes, while promoting resistance to standard treatments. New, personalised therapies that specifically address these molecular, genetic and immunological challenges are sorely needed.
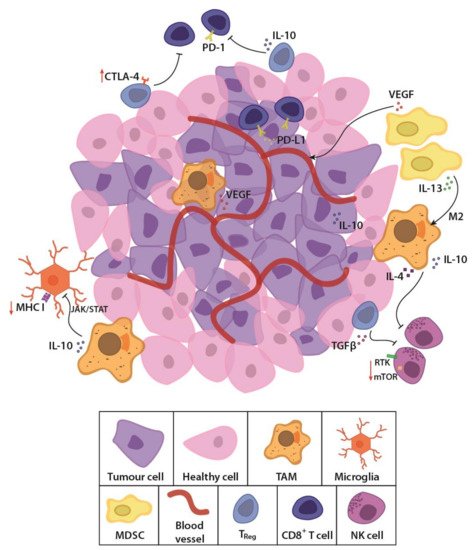
Figure 1. Schematic representation of the glioblastoma (GBM) tumour microenvironment. The evolving tumour microenvironment consists of diffusely infiltrative tumour cells with a leaky neovasculature, anti-inflammatory stromal cells and high concentrations of immunosuppressive cytokines and signalling molecules. Together, these factors generate a hostile environment where proliferation and persistence of cytotoxic immune cells are significantly hampered. TAM, tumour-associated macrophage; MDSC, myeloid derived suppressor cell; Treg, regulatory T cell; NK cell, natural killer cell; TGFβ, transforming growth factor β; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T lymphocyte-associated protein-4; IL-13, interleukin-13; IL-10, interleukin-10; VEGF, vascular endothelial growth factor; MHC I, major histocompatibility complex class I; JAK/STAT, Janus kinase/signal transducers and activators of transcription; RTK, receptor tyrosine kinase; mTOR, mammalian target of rapamycin.
2. Challenging Solid Brain Tumours with Natural Killers
Another approach recently translated into clinical trials is chimeric antigen receptor (CAR) NK therapy. CARs are a synthetic, modified representation of the TCR complex, that contains both an antigen-binding site and a T cell costimulatory domain that allows for autonomous, MHC-independent activation of the T cells [27][28]. They consist of an extracellular single-chain variable fragment (ScFv) derived from an antibody, and an intracellular domain with signalling motifs. Over time, adjustments made to CARs have targeted their structure; where first-generation CARs contained the intracellular CD3ζ from a TCR, second-generation CARs included the costimulatory CD28 or 4-1BB endodomains fused to CD3ζ (reviewed in [29]). Originally, the only difference between CAR T and CAR NK cells was the lymphocyte that held the CAR. The CARs had the same structure in both lymphocytes, since CARs with the CD3ζ chain alone or in combination with other costimulatory domains also promoted NK cell activation [30]. However, as the field advanced, domains from adaptor molecules associated with NK receptors such as DAP10 and DAP12 have been introduced. While CAR NK cells with a DAP10 domain showed poor efficiency [31], CAR NK cells with DAP12 demonstrated enhanced cytotoxicity when compared with those with CD3ζ [32].
Cytotoxic T cells and NK cells differ in many ways. Unlike T cells, NK cells are innate lymphocytes that specialise in recognising and killing transformed- and virus-infected cells through germline-encoded receptors that yield activating or inhibiting signals when interacting with target cell surface ligands [33][34][35]. Activating receptors include the natural cytotoxicity receptors NKp30 [36], NKp44 [37] and NKp46 [38], NK group 2 D (NKG2D) receptor [39], and killer immunoglobulin-like receptors (KIRs) with short cytoplasmic domains [40][41][42]. With these receptors, educated, cytotoxic CD56dim NK cells can become activated and kill tumour cells through a variety of mechanisms [43][44]. We have shown that NK cells that selectively express particular KIRs potently kill GBM cells and GBM stem-like cells [42][45][46]. A recent study showed enhanced killing capability of NK cells against GBM stem-like cells when inhibiting αv integrin or TGFβ [47]. Unlike CAR T cells, in the event of antigen loss by the tumour cells, CAR NK cells may still possess the ability to kill by their intrinsic activating receptors [48][49][50][51][52]. In addition, CAR NK cells are considered safer than CAR T cells since they are not known to induce immune effector cell-associated neurotoxicity syndrome (ICANS) or cytokine release syndrome (CRS) [53][54][55].
Importantly, allogeneic T cells are known to induce graft-versus-host disease (GvHD) while NK cells do not [53]. Autologous T cell therapy may not be amenable for all patients since those with significant lymphopenia due to prior anti-cancer treatments or those with a rapidly progressive disease will not be eligible due to insufficient numbers of T cells to expand into therapeutic doses of CAR T cells. This is important for GBM, as both treated and treatment-naïve GBM patients experience lymphopenia [56]. The protracted time required for CAR T cell production is also a bottleneck [55]. The first CAR NK cell approaches focused on primary peripheral blood-derived NK cells transfected with the CAR construct [57], but NK cells are more difficult to transfect than T cells [58]. To circumvent difficulties in introducing gene edits to NK cells, a recent publication has outlined a method for efficient electroporation of CRISPR-associated Cas9 ribonuclear protein (RNP) complexes in primary NK cells [59]. A similar protocol has been used to efficiently knock out the immune checkpoint receptor T-cell immunoglobulin mucin family member 3 (TIM3) in primary NK cells from healthy donors [60], PD-1 [61], and CD38 [62]. However, NK cells only represent approximately 5–15% of circulating lymphocytes in healthy adult humans [63] and require extensive ex vivo expansion. Additionally, they have a much shorter life span than T cells [64], although the persistence of CD-19-directed, IL-15 expressing CAR NK cells from cord blood were shown to survive for at least 4 weeks in mouse models [65]. In humans, multiple or higher doses of engineered NK cells might be required for a sustained treatment effect. NK cells may be obtained from various tissue sources, however, such as umbilical cord blood, induced pluripotent stem cells (iPSCs) or cell lines, such as NK-92 [66]. This characteristic provides the opportunity for “off-the-shelf” cells that can be applied to any patient, which is less readily achieved with autologous T cell therapies [58]. However, the possibility of a host-versus-graft scenario, where the implanted cells are systematically destroyed by the host immune system, must also be considered. Research into the production of reliable, “off-the-shelf” alternatives for cancer immunotherapy is currently in progress for both T and NK cells (reviewed in [67][68]). Strategies to avoid GvHD include knockout of the TCR in engineered T cells [69][70], while host attacks and graft rejection may be mitigated through knockout of MHC or application of lymphodepleting chemotherapeutics prior to engineered T/NK cell administration [71][72][73][74]. In GBM, NK cells have been shown to comprise only a small portion of immune cells in the tumour microenvironment, and to have a CD56+ CD16- phenotype [12], indicating that these cells may not be capable of killing CAR-expressing cells. Another opportunity may lie in NK cells’ inherent expression of KIR2DL4, which can bind HLA G to inhibit host NK cells [75][76].
The heterogeneity of GBM creates a significant challenge for the identification of a ubiquitously expressed tumour specific target for CAR constructs. In addition, GBM cells grown in stem cell media, which most closely preserve the original tumour phenotype, tend to exhibit reduced expression of both classical and non-classical MHC class I ligands for inhibitory NK receptors [12][77]. Therefore, these conditions, if present within patient tumour microenvironments, may provide additional opportunities for receptor mediated cytotoxicity in CAR NK cells [78][79] via both KIR and natural cytotoxicity receptors against stress-induced ligands known to be expressed by GBM tumour cells [45][46][80]. Bispecific TanCAR NK cells could be used to mitigate possible tumour escape by targeting two antigens [81]. As such, the choice of either CAR T or NK cell therapy depends on the strategy utilised, as the two provide different advantages and disadvantages, as summarised in Table 1. Taken together, CAR NK cells represent potentially superior effectors for solid tumours such as GBM.
Table 1. Comparisons between CAR T and NK cell therapies.
|
Comparisons to Consider |
CAR T |
CAR NK |
|---|---|---|
| Cell source | Autologous setting | Allogeneic setting or cell lines [66] |
| Off-the-shelf | Not eligible | Eligible [58] |
| GvHD | Induces GvHD | Not known to induce GvHD |
| Gene delivery | Effective by transfection or transduction | Effective by Cas9 RNP |
| Patients as candidates | Restricted to patient’s condition | All patients are potential candidates |
| Off-target effects | ICANS/CRS [82][83] | No/minimal ICANS or CRS |
| Response to antigen loss | No alternative mechanism | Response by existing NK cell receptors |
| Infusion dose | Single/few doses | Multiple or high |
| In vivo persistency | Longer (several months) [84] | Shorter (days to weeks) [64] |
| FDA/EMA approval | Approved [85][86] | Not yet approved |
| Cancer indication | Haematological malignancies [87] | Alternative for solid tumours |
GvHD, graft versus host disease; RNP, ribonuclear protein; ICANS, immune effector cell-associated neurotoxicity syndrome; CRS, cytokine release syndrome; FDA, U.S. Food and Drug Administration; EMA, European Medicines Agency.
In Vitro and In Vivo Clinical Studies for Designer NK Cells in GBM
The preclinical demonstration of the anti-tumour efficacy of CAR NK cells in haematological malignancies [31][88] has stimulated a number of clinical trials involving patients with these cancers (reviewed in [89]). Some CAR NK cell approaches proposed for the treatment of GBM have also demonstrated anti-tumour efficacy and extended animal survival in preclinical settings. Murakami et al. established a CAR KHYG-1 NK cell line which exhibited capacity for killing epidermal growth factor receptor (EGFR) variant III (EGFRvIII)-expressing GBM cells in vitro [90]. However, it lacked in vivo anti-tumour efficacy in mice ectopically injected with GBM cells [91]. Genßler et al. and Han et al. successfully generated CAR NK-92 cells bispecific for wild type EGFR and EGFRvIII and demonstrated their efficacy in GBM both in vitro and in vivo, when injected intracranially in GBM-bearing mice [92][93]. Furthermore, Zhang et al. showed the efficacy of another NK-92 CAR cell line, the NK-92/5.28.z, which is specific for human epidermal growth factor receptor 2 (HER2)/ErbB2, against GBM both in vitro and in vivo, through intratumoural injections into the brain [94]. Interestingly, these are the only CAR NK cell therapy constructs being tested in clinical trials for human GBM (clinical trial ID NCT03383978, CAR2BRAIN), although other avenues for CAR NK in GBM are in development (reviewed in [95]). The CAR2BRAIN study is in phase I, aiming to be completed by December 2022, with the primary goals of assessing safety and tolerability, persistence of NK-95/5.28.z cells and cytokine profile in cerebrospinal fluid and blood for 24 weeks after injection. The patients undergoing this trial receive only one injection of the engineered cells, and so far, no dose-limiting effects have been reported. Nevertheless, it remains to be seen if this and future trials will have to incorporate more doses to see a sustained benefit.
3. Picking a Suitable Target for GBM Immunotherapy
In GBM, heterogenous mutations are typically not immunogenic [96][97], which renders the tumours resistant to new generation immunotherapies, including CAR T and immune checkpoint blockade (reviewed in [98][99]). Choosing one or more suitable targets is critical for a sustained, beneficial effect. In addition to absent or limited expression in healthy tissues, the target antigen must have persistent expression on the tumour cells and present an appropriate affinity to the CAR which allows recognition of the target with low off-target toxicity [100]. An overview of immunotherapeutic targets for the treatment of GBM currently in clinical trials is shown in Table 2, which includes EGFRvIII, HER2/ErbB2, interleukin-13 receptor alpha 2 (IL13Rα2), B7H3 and NKG2D ligands. Although T cell exhaustion may be limited by targeting CTLA-4 and PD-1, chemotherapeutics with monoclonal antibodies against CTLA-4, PD-1 and PD-L1 have shown mixed and unpredictable response in GBM [101]. In a recent publication, however, Cloughesy et al. performed a randomised, early phase clinical trial using neoadjuvant pembrolizumab, a PD-1 inhibitor, which showed both overall and progression-free survival benefit to patients with recurrent GBM when applied both before and after surgery [102]. These results emphasize the importance of timing of treatment.
Table 2. Current targets used in CAR T and NK cell therapy for GBM.
| Clinical Trial ID | Target | Cell Type/Expression | Trial Method | Trial Status | Reference |
|---|---|---|---|---|---|
| NCT03726515, NCT01454596 | EGFRvIII | Glioma cells, 64% of GBM cases | CAR T | Completed, completed | [103] |
| NCT03383978, NCT01109095 |
HER2/ErbB2 | Tumour cells, normal endothelial and neuronal brain cells (low expression) | CAR NK | Recruiting, completed | [104] |
| NCT04003649, NCT02208362 | IL13Rα2 | Tumour cells, TAMs, MDSCs, overexpressed in 50% of GBM cases | CAR T | Recruiting, recruiting | [103] |
| NCT04077866, NCT04385173 | B7H3 | DC, T cells, overexpression on GBM tumour cells | CAR T | Recruiting, recruiting | [105] |
| NCT04270461 | NKG2DL | Receptor expressed on NK, NKT and T cells, ligands expressed on tumour cells | CAR T | Withdrawn | [48] |
EGFRvIII, epidermal growth factor receptor variant III; HER2, human epidermal growth factor receptor 2; ErbB2, Erb-B2 Receptor Tyrosine Kinase 2; IL13Rα2, interleukin 13 receptor subunit alpha 2; B7H3, B7 homologue 3; NKG2D, natural killer group 2 member D; TAM, tumour-associated macrophage; MDSC, myeloid-derived suppressor cell; DC, dendritic cell; NKT, natural killer T cell.
3.1. Growth Factor Receptors as Targets
One of the initial targets for GBM immunotherapy trials with CAR T cells was EGFRvIII, a constitutively active mutated variant of the EGFR receptor. Currently, there are ten active CAR T treatments for GBM in clinical trials, two of which target EGFRvIII (clinical trial ID NCT01454596 and NCT03726515). Results from different studies are contradicting based on the independent prognostic value of EGFRvIII in GBM for overall survival of newly diagnosed patients with EGFRvIII positive tumours [106][107]. A phase I clinical trial reported that a single infusion of EGFRvIII-directed CAR-T cell led to loss of antigen, and induction of adaptive resistance through increased PD-L1, IDO1, IL-10 and the presence of FoxP3+ Treg cells [108]. These results highlight the problems of early antigen loss and swift immunosuppressive response in these patients. HER2/ErbB2 is structurally homologous to EGFR, and mRNA has been found in 76% of primary GBM cell lines [109]. However, its expression levels in normal brain tissue, especially neuronal and endothelial cells of the cerebral cortex [110], pose difficulties in developing immunotherapy options with tolerable side-effects for brain tumour application. Nevertheless, pre-clinical studies demonstrated HER2-specific T lymphocyte cytotoxicity against GBM stem-like cells that was not evident in HER2- cells [104]. In addition, HER2-directed CAR NK cells engineered from healthy donors and breast cancer patients have been shown to selectively kill HER2+ tumour cells while avoiding healthy cells in vitro [111]. HER2 is also currently the only target used for CAR NK cell therapy in human trials for GBM with the CAR2BRAIN study in Germany (clinical trial ID NCT03383978), as mentioned above.
3.2. Interleukin-13 Receptor Alpha 2
IL13Rα2 is another cancer-associated receptor currently used in CAR T cell therapy for GBM (clinical trial ID NCT04003649 and NCT02208362). Overexpression in GBM coupled with low to no detectable levels in healthy brain tissue render IL13Rα2 a suitable target for immunotherapy [103]. The native IL13Rα2 has been reported to induce an invasive phenotype in GBM by promoting epithelial-mesenchymal transition. However, through interaction with EGFRvIII, IL13Rα2 signalling induces proliferation through activation of the STAT3 pathway [112]. Thus, dual targeting of IL13Rα2 and EGFRvIII could be a promising strategy for GBM CAR therapy. One study has reported on the administration of IL13Rα2 CAR T cells intracranially post resection, followed by further infusions both intraventricular and intrathecally in a single patient [113]. Although the patient experienced disease recurrence after adoptive cell therapy, remarkable tumour shrinkage was observed in all lesions by 77–100%, exhibiting a sustained responses for 7.5 months after intrathecal administration.
3.3. Co-Stimulator B7H3
B7H3 (CD276) is a costimulatory protein on the cell surface, where the B7 superfamily of proteins was initially thought to be solely involved in immune co-stimulation [114][115]. It has since been found to be highly expressed on GBM and tumour-associated endothelial cells, with protein levels increasing with increasing tumour grade [105][116]. Although its functional role is not fully elucidated, B7H3 is associated with poorer prognosis in GBM patients [116], making it a potential target for therapy. A recent clinical report using CAR-T cells directed towards B7H3 showed in one patient with recurrent GBM who received seven infusions significantly reduced volume of contrast enhancing lesion on MRI and clinical response for 50 days [117]. Two B7H3-directed CAR T cell clinical trials for GBM are currently in recruitment in China (clinical trial ID NCT04077866 and NCT04385173).
3.4. Multitarget Approach with NKG2D
The TAAs mentioned above are all considered feasible targets for GBM immunotherapy. However, their heterogenous expression on cancer and normal cells present challenges with toxicity, antigen escape and subsequent recurrence. One suggested approach to overcome this issue has been to use the lectin-like, type 2 transmembrane receptor NKG2D in CAR therapy. NKG2D is an activating receptor expressed on the surface of NK and CD8+ T cells [12][118][119] that binds MHC class-related chains A and B (MICA and MICB, respectively), as well as UL16-binding protein 6 (ULBP6), that are typically only expressed during hyper-proliferation and transformation [120]. NKG2D ligands are expressed by stressed and transformed cells, potentially induced as a response to chemo- and radiotherapy [39][121]. In immunocompetent mice, a recent study reported that an NKG2D-based CAR T therapy worked synergistically with radiotherapy, yielding persistence and long-term protection from disease [122]. Weiss et al. incorporated the full-length NKG2D protein fused to CD3ζ in association with DAP10, functioning similarly to a second-generation CAR, and transduced it into T cells. These promising results provide evidence for clinical translation into human trials. However, the single approved NKG2D-based CAR T clinical trial (clinical trial ID NCT04270461) has since been withdrawn due to administrative reasons.
4. Genetic Modification as an Aid to Existing Therapies
Cutting edge gene editing technologies [123] provide the opportunity to enhance the efficacy of existing molecular immunotherapy strategies. Introduction of clustered regularly interspaced short palindromic repeats (CRISPR) and the CRISPR-associated endonuclease Cas (typically Cas9) to design T or NK cells may improve the potency of cancer immunotherapy. CRISPR/Cas9 technology is used to knockout genes by inducing double-stranded DNA breaks (DSB) at specific sites within the genome. The Cas9 enzyme forms a complex with a 20-base guide RNA (gRNA) that is predesigned to recognize a complementary DNA target site in a gene of interest. Cas9 locates a protospacer adjacent motif (PAM) sequence directly downstream of the gRNA sequence, binds and triggers a DSB at the genomic target site [124] (Figure 3). The cell then repairs the damage through mechanisms based on the cell cycle stage and proliferative status [125][126]. The most common repair pathway is non-homologous end joining, wherein the two ends of the DNA are fused, often resulting in faulty repair, with random insertion or deletion of nucleotides causing frameshift mutations and thus a knockout of the gene. The other, less common pathway, homology-directed repair (HDR), uses a DNA repair template to repair the damaged DNA, or introduces a gene edit, as with HDR CRISPR [127]. HDR CRISPR, however, is difficult to accomplish and often leads to high numbers of unwanted insertions and deletions (indels) outside the genetic area of interest, so-called off-target effects [128][129].
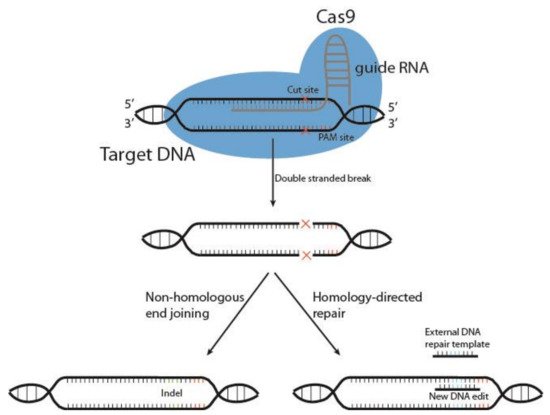
Figure 2. Schematic representation of CRISPR/Cas9 gene editing. The DNA endonuclease Cas9 is introduced to target cells via transduction, transfection or addition of pure Cas9 protein, and complexes with the guide RNA (gRNA). This complex between Cas9 and gRNA searches for a protospacer adjacent motif (PAM) sequence in proximity to the gRNA 20-base sequence. Once bound, Cas9 causes a double-stranded DNA break, and the cell must use either non-homologous end joining or homology-directed repair to fix the break. This results in either wild type DNA, edited DNA, or insertions and deletions (indels) that result in gene knockout.
The use of CRISPR/Cas9 technology in the development of cancer therapy has only recently begun to have an impact on human trials, as the ethical use of CRISPR/Cas9 to edit the human genome has been a hotly debated topic [130][131]. CRISPR/Cas9 mutagenesis screening has been used as a tool to identify REGNASE-1 as a negative regulator of antitumour responses, whose knockout improved efficiency and persistence of effector T cells in vitro [132]. In addition, CRISPR/Cas9 was used to successfully generate CBLB knockout NK cells derived from human placental stem-cells, without altering their phenotype [133], indicating possibility for use in primary lymphocytes. Preclinical research has successfully used multiplex CRISPR/Cas9 to limit universal CAR T cell exhaustion by disrupting the PD-1 gene, as well as the endogenous TCR α chain (TRAC) and β-2 microglobulin (B2M) [70]. The T cells, directed against EGFRvIII, were administered intracerebrally in GBM bearing mice, and enhanced survival. In a recent phase I human clinical trial, CRISPR/Cas9 was used on autologous T cells to knock out the endogenous TCR α and β chains as well as PD-1 in 3 patients with refractory cancer: 2 patients with multiple myeloma, and 1 with myxoid and round cell liposarcoma [69]. In addition, a transgenic TCR was transduced into the cells, targeting the cancer testis antigen NY-ESO-1. The T cells were then heterogeneously expanded and intravenously reinfused into the patients. The engineered cells persisted in the patients for up to 9 months. Stadtmauer et al. showed that at the time of infusion, the amount of Cas9 protein had diminished to a non-detectable level in the engineered T cells, had acceptable levels of off-target editing, and that the patients did not develop humoral responses to Cas9.
References
- Burnet, F.M. The concept of immunological surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27.
- Shi, L.; Kam, C.M.; Powers, J.C.; Aebersold, R.; Greenberg, A.H. Purification of three cytotoxic lymphocyte granule serine proteases that induce apoptosis through distinct substrate and target cell interactions. J. Exp. Med. 1992, 176, 1521–1529.
- Shi, L.; Kraut, R.P.; Aebersold, R.; Greenberg, A.H. A natural killer cell granule protein that induces DNA fragmentation and, apoptosis. J. Exp. Med. 1992, 175, 553–566.
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649.
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10.
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594.
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034.
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266.
- Tindemans, I.; Serafini, N.; Di Santo, J.P.; Hendriks, R.W. GATA-3 function in innate and adaptive immunity. Immunity 2014, 41, 191–206.
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186.
- Bahador, M.; Gras Navarro, A.; Rahman, M.A.; Dominguez-Valentin, M.; Sarowar, S.; Ulvestad, E.; Njølstad, G.; Lie, S.A.; Kristoffersen, E.K.; Bratland, E.; et al. Increased infiltration and tolerised antigen-specific CD8(+) T(EM) cells in tumor but not peripheral blood have no impact on survival of HCMV(+) glioblastoma patients. Oncoimmunology 2017, 6, e1336272.
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83.
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12.
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Pearson, J.R.D.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 2020, 11, 582106.
- Moserle, L.; Casanovas, O. Anti-angiogenesis and metastasis: A tumour and stromal cell alliance. J. Intern. Med. 2013, 273, 128–137.
- Rampling, R.; Cruickshank, G.; Lewis, A.D.; Fitzsimmons, S.A.; Workman, P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 427–431.
- Collingridge, D.R.; Piepmeier, J.M.; Rockwell, S.; Knisely, J.P. Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother. Oncol. 1999, 53, 127–131.
- Evans, S.M.; Jenkins, K.W.; Chen, H.I.; Jenkins, W.T.; Judy, K.D.; Hwang, W.T.; Lustig, R.A.; Judkins, A.R.; Grady, M.S.; Hahn, S.M.; et al. The Relationship among Hypoxia, Proliferation, and Outcome in Patients with De Novo Glioblastoma: A Pilot Study. Transl. Oncol. 2010, 3, 160–169.
- Hutchins, A.P.; Diez, D.; Miranda-Saavedra, D. The IL-10/STAT3-mediated anti-inflammatory response: Recent developments and future challenges. Brief. Funct. Genom. 2013, 12, 489–498.
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2021, 2, 723–740.
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.-E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955.
- Hao, C.; Chen, G.; Zhao, H.; Li, Y.; Chen, J.; Zhang, H.; Li, S.; Zhao, Y.; Chen, F.; Li, W.; et al. PD-L1 Expression in Glioblastoma, the Clinical and Prognostic Significance: A Systematic Literature Review and Meta-Analysis. Front. Oncol. 2020, 10.
- Bergmann, N.; Delbridge, C.; Gempt, J.; Feuchtinger, A.; Walch, A.; Schirmer, L.; Bunk, W.; Aschenbrenner, T.; Liesche-Starnecker, F.; Schlegel, J. The Intratumoral Heterogeneity Reflects the Intertumoral Subtypes of Glioblastoma Multiforme: A Regional Immunohistochemistry Analysis. Front. Oncol. 2020, 10, 494.
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410.
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968.
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028.
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049.
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283.
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383.
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J. Immunol. 2015, 194, 3201–3212.
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121.
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239.
- Lanier, L.L. Natural killer cell receptor signaling. Curr. Opin. Immunol. 2003, 15, 308–314.
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516.
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072.
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular cloning of NKp46: A novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998, 188, 953–960.
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441.
- Moretta, A.; Biassoni, R.; Bottino, C.; Pende, D.; Vitale, M.; Poggi, A.; Mingari, M.C.; Moretta, L. Major histocompatibility complex class I-specific receptors on human natural killer and T lymphocytes. Immunol. Rev. 1997, 155, 105–117.
- Campbell, K.S.; Purdy, A.K. Structure/function of human killer cell immunoglobulin-like receptors: Lessons from polymorphisms, evolution, crystal structures and mutations. Immunology 2011, 132, 315–325.
- Poggi, A.; Pella, N.; Morelli, L.; Spada, F.; Revello, V.; Sivori, S.; Augugliaro, R.; Moretta, L.; Moretta, A. p40, a novel surface molecule involved in the regulation of the non-major histocompatibility complex-restricted cytolytic activity in humans. Eur. J. Immunol. 1995, 25, 369–376.
- Ljunggren, H.G.; Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244.
- Lanier, L.L.; Yu, G.; Phillips, J.H. Co-association of CD3 zeta with a receptor (CD16) for IgG Fc on human natural killer cells. Nature 1989, 342, 803–805.
- Haspels, H.N.; Rahman, M.A.; Joseph, J.V.; Navarro, A.G.; Chekenya, M. Glioblastoma stem-like cells are more susceptible than differentiated cells to natural killer cell lysis mediated through killer immunoglobulin-like receptors-human leukocyte antigen ligand mismatch and activation receptor-ligand interactions. Front. Immunol. 2018, 9.
- Gras Navarro, A.; Kmiecik, J.; Leiss, L.; Zelkowski, M.; Engelsen, A.; Bruserud, Ø.; Zimmer, J.; Enger, P.Ø.; Chekenya, M. NK Cells with KIR2DS2 Immunogenotype Have a Functional Activation Advantage To Efficiently Kill Glioblastoma and Prolong Animal Survival. J. Immunol. 2014, 193, 6192–6206.
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131.
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582.
- Souza-Fonseca-Guimaraes, F.; Cursons, J.; Huntington, N.D. The Emergence of Natural Killer Cells as a Major Target in Cancer Immunotherapy. Trends Immunol. 2019, 40, 142–158.
- Kärre, K. Natural killer cell recognition of missing self. Nat. Immunol. 2008, 9, 477–480.
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci. Transl. Med. 2020, 12.
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54.
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553.
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and biological correlates of neurotoxicity associated with car t-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018, 8, 958–971.
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876.
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468.
- Lin, C.Y.; Gobius, I.; Souza-Fonseca-Guimaraes, F. Natural killer cell engineering—A new hope for cancer immunotherapy. Semin. Hematol. 2020, 57, 194–200.
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781.
- Rautela, J.; Surgenor, E.; Huntington, N.D. Drug target validation in primary human natural killer cells using CRISPR RNP. J. Leukoc. Biol. 2020, 108, 1397–1408.
- Morimoto, T.; Nakazawa, T.; Matsuda, R.; Nishimura, F.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Park, Y.S.; Tsujimura, T.; Nakase, H. CRISPR-Cas9-Mediated TIM3 Knockout in Human Natural Killer Cells Enhances Growth Inhibitory Effects on Human Glioma Cells. Int. J. Mol. Sci. 2021, 22, 3489.
- Teratake, Y.; Takashina, T.; Iijima, K.; Sakuma, T.; Yamamoto, T.; Ishizaka, Y. Development of a protein-based system for transient epigenetic repression of immune checkpoint molecule and enhancement of antitumour activity of natural killer cells. Br. J. Cancer 2020, 122, 823–834.
- Gurney, M.; Stikvoort, A.; Nolan, E.; Kirkham-McCarthy, L.; Khoruzhenko, S.; Shivakumar, R.; Zweegman, S.; Van de Donk, N.; Mutis, T.; Szegezdi, E.; et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica 2020. Available online: https://haematologica.org/article/view/haematol.2020.271908 (accessed on 2 October 2021).
- Angelo, L.S.; Banerjee, P.P.; Monaco-Shawver, L.; Rosen, J.B.; Makedonas, G.; Forbes, L.R.; Mace, E.M.; Orange, J.S. Practical NK cell phenotyping and variability in healthy adults. Immunol. Res. 2015, 62, 341–356.
- Zhang, Y.; Wallace, D.L.; De Lara, C.M.; Ghattas, H.; Asquith, B.; Worth, A.; Griffin, G.E.; Taylor, G.P.; Tough, D.F.; Beverley, P.C.L.; et al. In vivo kinetics of human natural killer cells: The effects of ageing and acute and chronic viral infection. Immunology 2007, 121, 258–265.
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531.
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510.
- Mo, F.; Mamonkin, M.; Brenner, M.K.; Heslop, H.E. Taking T-Cell Oncotherapy Off-the-Shelf. Trends Immunol. 2021, 42, 261–272.
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965.
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367.
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304.
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266.
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349.
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894.
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864.
- Rajagopalan, S.; Long, E.O. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J. Exp. Med. 1999, 189, 1093–1100.
- Faure, M.; Long, E.O. KIR2DL4 (CD158d), an NK cell-activating receptor with inhibitory potential. J. Immunol. 2002, 168, 6208–6214.
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK Cells Recognize and Kill Human Glioblastoma Cells with Stem Cell-Like Properties. J. Immunol. 2009, 182, 3530–3539.
- Mostafa, H.; Pala, A.; Hogel, J.; Hlavac, M.; Dietrich, E.; Westhoff, M.A.; Nonnenmacher, L.; Burster, T.; Georgieff, M.; Wirtz, C.R.; et al. Immune phenotypes predict survival in patients with glioblastoma multiforme. J. Hematol. Oncol. 2016, 9, 77.
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341.
- Dominguez-Valentin, M.; Navarro, A.G.; Rahman, A.M.; Kumar, S.; Retière, C.; Ulvestad, E.; Kristensen, V.; Lund-Johansen, M.; Lie, B.A.; Enger, P.Ø.; et al. Identification of a natural killer cell receptor allele that prolongs survival of cytomegalovirus-positive glioblastoma patients. Cancer Res. 2016, 76, 5326–5336.
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2013, 2.
- Tallantyre, E.C.; Evans, N.A.; Parry-Jones, J.; Morgan, M.P.G.; Jones, C.H.; Ingram, W. Neurological updates: Neurological complications of CAR-T therapy. J. Neurol. 2021, 268, 1544–1554.
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Ann. Intensive Care 2017, 45, 2.
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056.
- Bouchkouj, N.; Kasamon, Y.L.; Claro, R.A.d.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA approval summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708.
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146.
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517.
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273.
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. eBioMedicine 2020, 59, 102975.
- Murakami, T.; Nakazawa, T.; Natsume, A.; Nishimura, F.; Nakamura, M.; Matsuda, R.; Omoto, K.; Tanaka, Y.; Shida, Y.; Park, Y.S.; et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. 2018, 38, 5049–5056.
- Nakazawa, T.; Murakami, T.; Natsume, A.; Nishimura, F.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Park, Y.-S.; et al. KHYG-1 Cells With EGFRvIII-specific CAR Induced a Pseudoprogression-like Feature in Subcutaneous Tumours Derived from Glioblastoma-like Cells. Anticancer Res. 2020, 40, 3231–3237.
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015, 5.
- Genßler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. OncoImmunology 2016, 5.
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2015, 108.
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol. 2021, 151, 41–53.
- Razavi, S.-M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3.
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.M.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7.
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794.
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486.
- Saikali, S.; Avril, T.; Collet, B.; Hamlat, A.; Bansard, J.Y.; Drenou, B.; Guegan, Y.; Quillien, V. Expression of nine tumour antigens in a series of human glioblastoma multiforme: Interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J. Neurooncol. 2007, 81, 139–148.
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485.
- Zhang, J.; Wang, J.; Marzese, D.M.; Wang, X.; Yang, Z.; Li, C.; Zhang, H.; Zhang, J.; Chen, C.C.; Kelly, D.F.; et al. B7H3 regulates differentiation and serves as a potential biomarker and theranostic target for human glioblastoma. Lab. Investig. 2019, 99, 1117–1129.
- Montano, N.; Cenci, T.; Martini, M.; D’Alessandris, Q.G.; Pelacchi, F.; Ricci-Vitiani, L.; Maira, G.; Maria, R.D.; Larocca, L.M.; Pallini, R. Expression of EGFRvIII in Glioblastoma: Prognostic Significance Revisited. Neoplasia 2011, 13, 1113–1121.
- Tripathy, K.; Das, B.; Singh, A.K.; Misra, A.; Misra, S.; Misra, S.S. Prognostic Significance of Epidermal Growth Factor Receptor in Patients of Glioblastoma Multiforme. J. Clin. Diagn. Res. 2017, 11, EC05–EC08.
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9.
- Liu, G.; Ying, H.; Zeng, G.; Wheeler, C.J.; Black, K.L.; Yu, J.S. HER-2, gp100, and MAGE-1 Are Expressed in Human Glioblastoma and Recognized by Cytotoxic T Cells. Cancer Res. 2004, 64.
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347.
- Portillo, A.L.; Hogg, R.; Poznanski, S.M.; Rojas, E.A.; Cashell, N.J.; Hammill, J.A.; Chew, M.V.; Shenouda, M.M.; Ritchie, T.M.; Cao, Q.T.; et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience 2021, 24, 102619.
- Newman, J.P.; Wang, G.Y.; Arima, K.; Guan, S.P.; Waters, M.R.; Cavenee, W.K.; Pan, E.; Aliwarga, E.; Chong, S.T.; Kok, C.Y.L.; et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nat. Commun. 2017, 8, 1913.
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569.
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-γ production. Nat. Immunol. 2001, 2, 269–274.
- Prasad, D.V.R.; Nguyen, T.; Li, Z.; Yang, Y.; Duong, J.; Wang, Y.; Dong, C. Murine B7-H3 Is a Negative Regulator of T Cells. J. Immunol. 2004, 173, 2500–2506.
- Lemke, D.; Pfenning, P.N.; Sahm, F.; Klein, A.C.; Kempf, T.; Warnken, U.; Schnolzer, M.; Tudoran, R.; Weller, M.; Platten, M.; et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin. Cancer Res. 2012, 18, 105–117.
- Tang, X.; Wang, Y.; Huang, J.; Zhang, Z.; Liu, F.; Xu, J.; Guo, G.; Wang, W.; Tong, A.; Zhou, L. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduct. Target 2021, 6, 125.
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress- inducible MICA. Science 1999, 285, 727–729.
- Wu, J.; Song, Y.; Bakker, A.B.H.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732.
- Lee, J.; Zhang, T.; Hwang, I.; Kim, A.; Nitschke, L.; Kim, M.; Scott, J.M.; Kamimura, Y.; Lanier, L.L.; Kim, S. Epigenetic Modification and Antibody-Dependent Expansion of Memory-like NK Cells in Human Cytomegalovirus-Infected Individuals. Immunity 2015, 42, 431–442.
- Weiss, T.; Schneider, H.; Silginer, M.; Steinle, A.; Pruschy, M.; Polic, B.; Weller, M.; Roth, P. NKG2D-Dependent antitumor effects of chemotherapy and radiotherapy against glioblastoma. Clin. Cancer Res. 2018, 24, 882–895.
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043.
- The Nobel Prize in Chemistry 2020. Available online: https://www.nobelprize.org/page/2/?np-keyword=press-release (accessed on 2 October 2021).
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71.
- Iliakis, G.; Wang, H.; Perrault, A.R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Panteliasc, G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004, 104, 14–20.
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139.
- Schiel, J.A.; Chou, E.; Mayer, M.; Anderson, E.M.; Van, A.; Smith, B. Homology-Directed Repair with Dharmacon™ Edit-R™ CRISPR-Cas9 Reagents and Single-Stranded DNA Oligos; Dharmacon, GE Healthcare: Buckinghamshire, UK, 2015.
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157.
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for improving HDR efficiency. Front Genet. 2019, 10, 691.
- Baltimore, D.; Berg, P.; Botchan, M.; Carroll, D.; Charo, R.A.; Church, G.; Corn, J.E.; Daley, G.Q.; Doudna, J.A.; Fenner, M.; et al. A prudent path forward for genomic engineering and germline gene modification. Science 2015, 348, 36–38.
- Lanphier, E.; Urnov, F. Don’t edit the human germ line. Nature 2015, 519, 410–411.
- Wei, J.; Long, L.; Zheng, W.; Dhungana, Y.; Lim, S.A.; Guy, C.; Wang, Y.; Wang, Y.D.; Qian, C.; Xu, B.; et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 2019, 576, 471–476.
- Guo, X.; Mahlakõiv, T.; Ye, Q.; Somanchi, S.; He, S.; Rana, H.; DiFiglia, A.; Gleason, J.; van der Touw, W.; Hariri, R.; et al. CBLB ablation with CRISPR/Cas9 enhances cytotoxicity of human placental stem cell-derived NK cells for cancer immunotherapy. J. Immunother. Cancer 2021, 9.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
922
Revisions:
3 times
(View History)
Update Date:
29 Mar 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No