 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giulia Spoto | + 4716 word(s) | 4716 | 2021-10-11 05:52:08 | | | |
| 2 | Beatrix Zheng | Meta information modification | 4716 | 2021-10-28 04:32:36 | | |
Video Upload Options
Seizures are the most frequent neurological clinical symptoms of the central nervous system (CNS) during the neonatal period. Neonatal seizures may be ascribed to an acute event or symptomatic conditions determined by genetic, metabolic or structural causes, outlining the so-called ‘Neonatal Epilepsies’. To date, three main groups of neonatal epilepsies are recognised during the neonatal period: benign familial neonatal epilepsy (BFNE), early myoclonic encephalopathy (EME) and ‘Ohtahara syndrome’ (OS).
1. Introduction
2. Epileptogenesis in the Neonatal Brain
3. Epileptic Phenotypes of Newborns
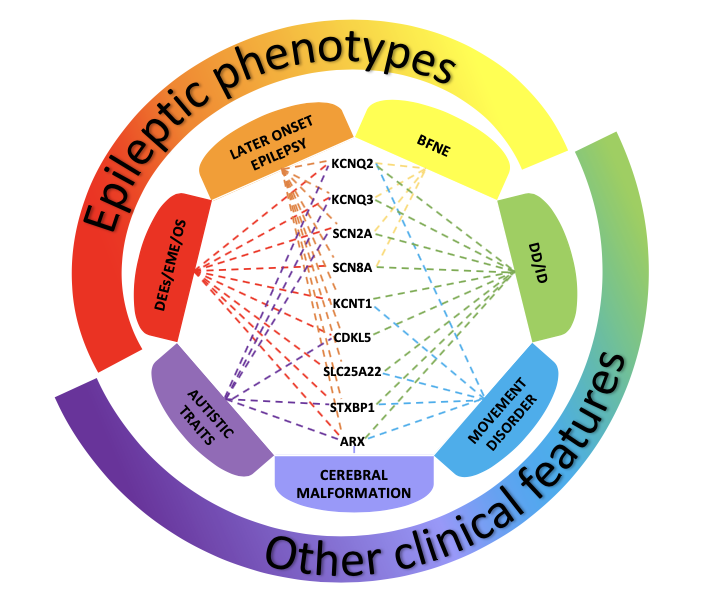
|
Figure 1. Disorders associated with variants of genes most frequently involved in neonatal-onset epilepsies The picture shows the clinical phenotypes associated with variants of genes most frequently involved in neonatal-onset epilepsies. Benign Familial Neonatal Epilepsy (BFNE) has been commonly linked to KCNQ2/3 genes, but also to SCN2A, and recently to a few cases of SCN8A variants. Pathogenic variants of all the reported genes have been related to severe forms of neonatal-onset developmental and epileptic encephalopathies (DEEs), such as Early Myoclonic Encephalopathy (EME) and Ohtahara syndrome (OS). Variants of these genes have also been associated with other clinical presentations, including later-onset epilepsy, developmental delay/intellectual disability (DD/ID), movement disorders, autistic traits and cerebral malformations. The overlap of these clinical features makes it difficult to establish a precise genotype-phenotype correlation, delaying the assessment of the condition and the start of a proper treatment. |
3.1. Benign Familial Neonatal Epilepsy (BFNE)
3.2. Developmental and Epileptic Encephalopathies (DEEs)
3.2.1. Early Myoclonic Encephalopathy (EME)/Ohtahara Syndrome (OS)
3.2.2. Genes and Pathogenic Variants Mostly Involved in DEEs
4. The Role of EEG in Neonatal Seizures
5. Treatment
6. Conclusion
References
- Shellhaas, R.A. Seizure classification, etiology, and management. Handb. Clin. Neurol. 2019, 162, 347–361.
- Pisani, F.; Percesepe, A.; Spagnoli, C. Genetic diagnosis in neonatal-onset epilepsies: Back to the future. Eur. J. Paediatr. Neurol. 2018, 22, 354–357.
- Glass, H.C.; Shellhaas, R.A.; Wusthoff, C.J.; Chang, T.; Abend, N.S.; Chu, C.J.; Cilio, M.R.; Glidden, D.V.; Bonifacio, S.L.; Massey, S.; et al. Neonatal Seizure Registry Study Group. Contemporary Profile of Seizures in Neonates: A Prospective Cohort Study. J. Pediatrics 2016, 174, 98–103.e1.
- Ramantani, G.; Schmitt, B.; Plecko, B.; Pressler, R.M.; Wohlrab, G.; Klebermass-Schrehof, K.; Hagmann, C.; Pisani, F.; Boylan, G.B. Neonatal Seizures-Are We there Yet? Neuropediatrics 2019, 50, 280–293.
- Katsarou, A.M.; Galanopoulou, A.S.; Moshé, S. Epileptogenesis in neonatal brain. Semin. Fetal Neonatal Med. 2018, 23, 159–167.
- Plouin, P.; Kaminska, A. Neonatal seizures. Handb. Clin. Neurol. 2013, 111, 467–476.
- Pressler, R.M.; Cilio, M.R.; Mizrahi, E.M.; Moshé, S.L.; Nunes, M.L.; Plouin, P.; Vanhatalo, S.; Yozawitz, E.; de Vries, L.S.; Puthenveettil Vinayan, K.; et al. The ILAE classification of seizures and the epilepsies: Modification for seizures in the neonate. Position Pap. By ILAE Task Force Neonatal Seizures. Epilepsia 2021, 62, 615–628.
- Okumura, A. Electroencephalography in neonatal epilepsies. Pediatrics Int. 2020, 62, 1019–1028.
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; van Emde Boas, W.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010, 51, 676–685.
- Spagnoli, C.; Fusco, C.; Percesepe, A.; Leuzzi, V.; Pisani, F. Genetic Neonatal-Onset Epilepsies and Developmental/Epileptic Encephalopathies with Movement Disorders: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 4202.
- Musto, E.; Gardella, E.; Møller, R.S. Recent advances in treatment of epilepsy-related sodium channelopathies. Eur. J. Paediatr. Neurol. 2020, 24, 123–128.
- Miao, P.; Tang, S.; Ye, J.; Wang, J.; Lou, Y.; Zhang, B.; Xu, X.; Chen, X.; Li, Y.; Feng, J. Electrophysiological features: The next precise step for SCN2A developmental epileptic encephalopathy. Mol. Genet. Genom. Med. 2020, 8, e1250.
- Oyrer, J.; Maljevic, S.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S.; Reid, C.A. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol. Rev. 2018, 70, 142–173.
- Bayat, A.; Bayat, M.; Rubboli, G.; Møller, R.S. Epilepsy Syndromes in the First Year of Life and Usefulness of Genetic Testing for Precision Therapy. Genes 2021, 12, 1051.
- Pitkänen, A. Therapeutic approaches to epileptogenesis—hope on the horizon. Epilepsia 2010, 51 (Suppl. S3), 2–17.
- Briggs, S.W.; Galanopoulou, A.S. Altered GABA signaling in early life epilepsies. Neural Plast. 2011, 2011, 527605.
- Galanopoulou, A.S. Sexually dimorphic expression of KCC2 and GABA function. Epilepsy Res. 2008, 80, 99–113.
- Galanopoulou, A.S. Mutations affecting GABAergic signaling in seizures and epilepsy. Pflug. Eur. J. Physiol. 2010, 460, 505–523.
- Galanopoulou, A.S.; Moshé, S. Pathogenesis and new candidate treatments for infantile spasms and early life epileptic encephalopathies: A view from preclinical studies. Neurobiol. Dis. 2015, 79, 135–149.
- EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 2014, 95, 360–370.
- Rakhade, S.N.; Jensen, F.E. Epileptogenesis in the immature brain: Emerging mechanisms. Nat. Rev. Neurol. 2009, 5, 380–391.
- Boylan, G.B.; Kharoshankaya, L.; Mathieson, S.R. Diagnosis of seizures and encephalopathy using conventional EEG and amplitude integrated EEG. Handb. Clin. Neurol. 2019, 162, 363–400.
- Solovieff, N.; Cotsapas, C.; Lee, P.H.; Purcell, S.M.; Smoller, J.W. Pleiotropy in complex traits: Challenges and strategies. Nat. Rev. Genet. 2013, 14, 483–495.
- McClellan, J.; King, M.C. Genetic heterogeneity in human disease. Cell 2010, 141, 210–217.
- Allen, N.M.; Weckhuysen, S.; Gorman, K.; King, M.D.; Lerche, H. Genetic potassium channel-associated epilepsies: Clinical review of the Kv family. Eur. J. Paediatr. Neurol. 2020, 24, 105–116.
- Vasudevan, C.; Levene, M. Epidemiology and aetiology of neonatal seizures. Semin. Fetal Neonatal Med. 2013, 18, 185–191.
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893.
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29.
- Howell, K.B.; McMahon, J.M.; Carvill, G.L.; Tambunan, D.; Mackay, M.T.; Rodriguez-Casero, V.; Webster, R.; Clark, D.; Freeman, J.L.; Calvert, S.; et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology 2015, 85, 958–966.
- Reynolds, C.; King, M.D.; Gorman, K.M. The phenotypic spectrum of SCN2A-related epilepsy. Eur. J. Paediatr. Neurol. 2020, 24, 117–122.
- Wolff, M.; Brunklaus, A.; Zuberi, S.M. Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia 2019, 60 (Suppl. S3), S59–S67.
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521.
- Olson, H.E.; Kelly, M.; LaCoursiere, C.M.; Pinsky, R.; Tambunan, D.; Shain, C.; Ramgopal, S.; Takeoka, M.; Libenson, M.H.; Julich, K.; et al. Genetics and genotype-phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann. Neurol. 2017, 81, 419–429.
- Milh, M.; Cacciagli, P.; Ravix, C.; Badens, C.; Lépine, A.; Villeneuve, N.; Villard, L. Severe neonatal seizures: From molecular diagnosis to precision therapy? Rev. Neurol. 2016, 172, 171–173.
- Ffrench-Constant, S.; Kachramanoglou, C.; Jones, B.; Basheer, N.; Syrmos, N.; Ganau, M.; Jan, W. Fetal and neonatal MRI features of ARX-related lissencephaly presenting with neonatal refractory seizure disorder. Quant. Imaging Med. Surg. 2019, 9, 1767–1772.
- Lammertse, H.; van Berkel, A.A.; Iacomino, M.; Toonen, R.F.; Striano, P.; Gambardella, A.; Verhage, M.; Zara, F. Homozygous STXBP1 variant causes encephalopathy and gain-of-function in synaptic transmission. Brain A J. Neurol. 2020, 143, 441–451.
- Saitsu, H.; Kato, M.; Mizuguchi, T.; Hamada, K.; Osaka, H.; Tohyama, J.; Uruno, K.; Kumada, S.; Nishiyama, K.; Nishimura, A.; et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 2008, 40, 782–788.
- Guiberson, N.; Pineda, A.; Abramov, D.; Kharel, P.; Carnazza, K.E.; Wragg, R.T.; Dittman, J.S.; Burré, J. Mechanism-based rescue of Munc18-1 dysfunction in varied encephalopathies by chemical chaperones. Nat. Commun. 2018, 9, 3986.
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflug. Eur. J. Physiol. 2004, 447, 689–709.
- Nicotera, A.G.; Dicanio, D.; Pironti, E.; Bonsignore, M.; Cafeo, A.; Efthymiou, S.; Mondello, P.; Salpietro, V.; Houlden, H.; Di Rosa, G. De novo mutation in SLC25A22 gene: Expansion of the clinical and electroencephalographic phenotype. J. Neurogenet. 2021, 35, 67–73.
- Gürsoy, S.; Erçal, D. Diagnostic Approach to Genetic Causes of Early-Onset Epileptic Encephalopathy. J. Child Neurol. 2016, 31, 523–532.
- Bodian, D.L.; Schreiber, J.M.; Vilboux, T.; Khromykh, A.; Hauser, N.S. Mutation in an alternative transcript of CDKL5 in a boy with early-onset seizures. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002360.
- Jakimiec, M.; Paprocka, J.; Śmigiel, R. CDKL5 Deficiency Disorder-A Complex Epileptic Encephalopathy. Brain Sci. 2020, 10, 107.
- Schroeder, E.; Yuan, L.; Seong, E.; Ligon, C.; DeKorver, N.; Gurumurthy, C.B.; Arikkath, J. Neuron-Type Specific Loss of CDKL5 Leads to Alterations in mTOR Signaling and Synaptic Markers. Mol. Neurobiol. 2019, 56, 4151–4162.
- Lim, C.X.; Ricos, M.G.; Dibbens, L.M.; Heron, S.E. KCNT1 mutations in seizure disorders: The phenotypic spectrum and functional effects. J. Med Genet. 2016, 53, 217–225.
- Numis, A.L.; Nair, U.; Datta, A.N.; Sands, T.T.; Oldham, M.S.; Patel, A.; Li, M.; Gazina, E.; Petrou, S.; Cilio, M.R. Lack of response to quinidine in KCNT1-related neonatal epilepsy. Epilepsia 2018, 59, 1889–1898.
- Kessi, M.; Chen, B.; Peng, J.; Tang, Y.; Olatoutou, E.; He, F.; Yang, L.; Yin, F. Intellectual Disability and Potassium Channelopathies: A Systematic Review. Front. Genet. 2020, 11, 614.
- Kim, H.J.; Yang, D.; Kim, S.H.; Kim, B.; Kim, H.D.; Lee, J.S.; Choi, J.R.; Lee, S.T.; Kang, H.C. Genetic and clinical features of SCN8A developmental and epileptic encephalopathy. Epilepsy Res. 2019, 158, 106222.
- Anand, G.; Collett-White, F.; Orsini, A.; Thomas, S.; Jayapal, S.; Trump, N.; Zaiwalla, Z.; Jayawant, S. Autosomal dominant SCN8A mutation with an unusually mild phenotype. Eur. J. Paediatr. Neurol. 2016, 20, 761–765.
- Johannesen, K.M.; Liu, Y.; Koko, M.; Gjerulfsen, C.E.; Sonnenberg, L.; Schubert, J.; Fenger, C.D.; Eltokhi, A.; Rannap, M.; Koch, N.A.; et al. Genotype-phenotype correlations in SCN8A-related disorders reveal prognostic and therapeutic implications. Brain A J. Neurol. 2021, awab321.
- Bye, A.M.; Flanagan, D. Spatial and temporal characteristics of neonatal seizures. Epilepsia 1995, 36, 1009–1016.
- Murray, D.M.; Boylan, G.B.; Ali, I.; Ryan, C.A.; Murphy, B.P.; Connolly, S. Defining the gap between electrographic seizure burden, clinical expression and staff recognition of neonatal seizures. Arch. Dis. Child.-Fetal Neonatal Ed. 2008, 93, F187–F191.
- Boylan, G.B.; Rennie, J.M.; Pressler, R.M.; Wilson, G.; Morton, M.; Binnie, C.D. Phenobarbitone, neonatal seizures, and video-EEG. Archives of disease in childhood. Fetal Neonatal Ed. 2002, 86, F165–F170.
- Scher, M.S.; Alvin, J.; Gaus, L.; Minnigh, B.; Painter, M.J. Uncoupling of EEG-clinical neonatal seizures after antiepileptic drug use. Pediatric Neurol. 2003, 28, 277–280.
- Cornet, M.C.; Morabito, V.; Lederer, D.; Glass, H.C.; Ferrao Santos, S.; Numis, A.L.; Ferriero, D.M.; Sands, T.T.; Cilio, M.R. Neonatal presentation of genetic epilepsies: Early differentiation from acute provoked seizures. Epilepsia 2021, 62, 1907–1920.
- Pressler, R.M.; Lagae, L. Why we urgently need improved seizure and epilepsy therapies for children and neonates. Neuropharmacology 2020, 170, 107854.
- Andrade, E.; Shaikh, Z.; Chavez, W.; Torres, A. Tratamiento de las convulsionesneonatales; Treatment of neonatal seizures. Medicina 2018, 78 (Suppl. S2), 30–35.
- Sharma, D.; Hussain, A.M.; Sharma, S.S. Efficacy of Levetiracetam in neonatal seizures: A systematic review. J. Matern. Fetal Neonatal Med. 2020, 1–8.
- Bittigau, P.; Sifringer, M.; Ikonomidou, C. Antiepileptic drugs and apoptosis in the developing brain. Ann. N. Y. Acad. Sci. 2003, 993, 103–124.
- Millichap, J.J.; Park, K.L.; Tsuchida, T.; Ben-Zeev, B.; Carmant, L.; Flamini, R.; Joshi, N.; Levisohn, P.M.; Marsh, E.; Nangia, S.; et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2016, 2, e96.
- Millichap, J.J.; Miceli, F.; De Maria, M.; Keator, C.; Joshi, N.; Tran, B.; Soldovieri, M.V.; Ambrosino, P.; Shashi, V.; Mikati, M.A.; et al. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia 2017, 58, e10–e15.
- Kuersten, M.; Tacke, M.; Gerstl, L.; Hoelz, H.; Stülpnagel, C.V.; Borggraefe, I. Antiepileptic therapy approaches in KCNQ2 related epilepsy: A systematic review. Eur. J. Med. Genet. 2020, 63, 103628.
- Fitzgerald, M.P.; Fiannacca, M.; Smith, D.M.; Gertler, T.S.; Gunning, B.; Syrbe, S.; Verbeek, N.; Stamberger, H.; Weckhuysen, S.; Ceulemans, B.; et al. Treatment Responsiveness in KCNT1-Related Epilepsy. Neurotherapeutics 2019, 16, 848–857.
- Dilena, R.; Striano, P.; Traverso, M.; Viri, M.; Cristofori, G.; Tadini, L.; Barbieri, S.; Romeo, A.; Zara, F. Dramatic effect of levetiracetam in early-onset epileptic encephalopathy due to STXBP1 mutation. Brain Dev. 2016, 38, 128–131.
- Li, T.; Cheng, M.; Wang, J.; Hong, S.; Li, M.; Liao, S.; Xie, L.; Jiang, L. De novo mutations of STXBP1 in Chinese children with early onset epileptic encephalopathy. Genes Brain Behav. 2018, 17, e12492.
- Bialer, M. New antiepileptic drugs that are second generation to existing antiepileptic drugs. Expert Opin. Investig. Drugs 2006, 15, 637–647.
- Matagne, A.; Margineanu, D.G.; Kenda, B.; Michel, P.; Klitgaard, H. Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. Br. J. Pharmacol. 2008, 154, 1662–1671.
- Schubert-Bast, S.; Willems, L.M.; Kurlemann, G.; Knake, S.; Müller-Schlüter, K.; Rosenow, F.; Strzelczyk, A. Postmarketing experience with brivaracetam in the treatment of focal epilepsy in children and adolescents. Epilepsy Behav. EB 2018, 89, 89–93.
- Pustorino, G.; Spano, M.; Sgro, D.L.; Di Rosa, G.; Tricomi, G.; Bellantone, D.; Tortorella, G. Status gelasticus associated with levetiracetam as add-on treatment. Epileptic Disord. Int. Epilepsy J. Videotape 2007, 9, 186–189.
- Gowda, V.K.; Nagarajan, B.; Shivappa, S.K.; Benakappa, N. Effectiveness and Safety of Brivaracetam in Children. Indian J. Pediatrics 2021, 88, 506.
- Nissenkorn, A.; Tzadok, M.; Bar-Yosef, O.; Ben-Zeev, B. Treatment with brivaracetam in children—The experience of a pediatric epilepsy center. Epilepsy Behav. EB 2019, 101 Pt A, 106541.
- Di Rosa, G.; Dicanio, D.; Nicotera, A.G.; Mondello, P.; Cannavò, L.; Gitto, E. Efficacy of Intravenous Hydrocortisone Treatment in Refractory Neonatal Seizures: A Report on Three Cases. Brain Sci. 2020, 10, 885.

