 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniel Sausen | + 3474 word(s) | 3474 | 2021-07-22 05:17:34 | | | |
| 2 | Peter Tang | Meta information modification | 3474 | 2021-10-27 02:58:10 | | |
Video Upload Options
The human immune system boasts a diverse array of strategies for recognizing and eradicating invading pathogens. Human betaherpesviruses, a highly prevalent subfamily of viruses, include human cytomegalovirus (HCMV), human herpesvirus (HHV) 6A, HHV-6B, and HHV-7. These viruses have evolved numerous mechanisms for evading the host response.
1. Introduction
2. Innate Immune Response Pathways
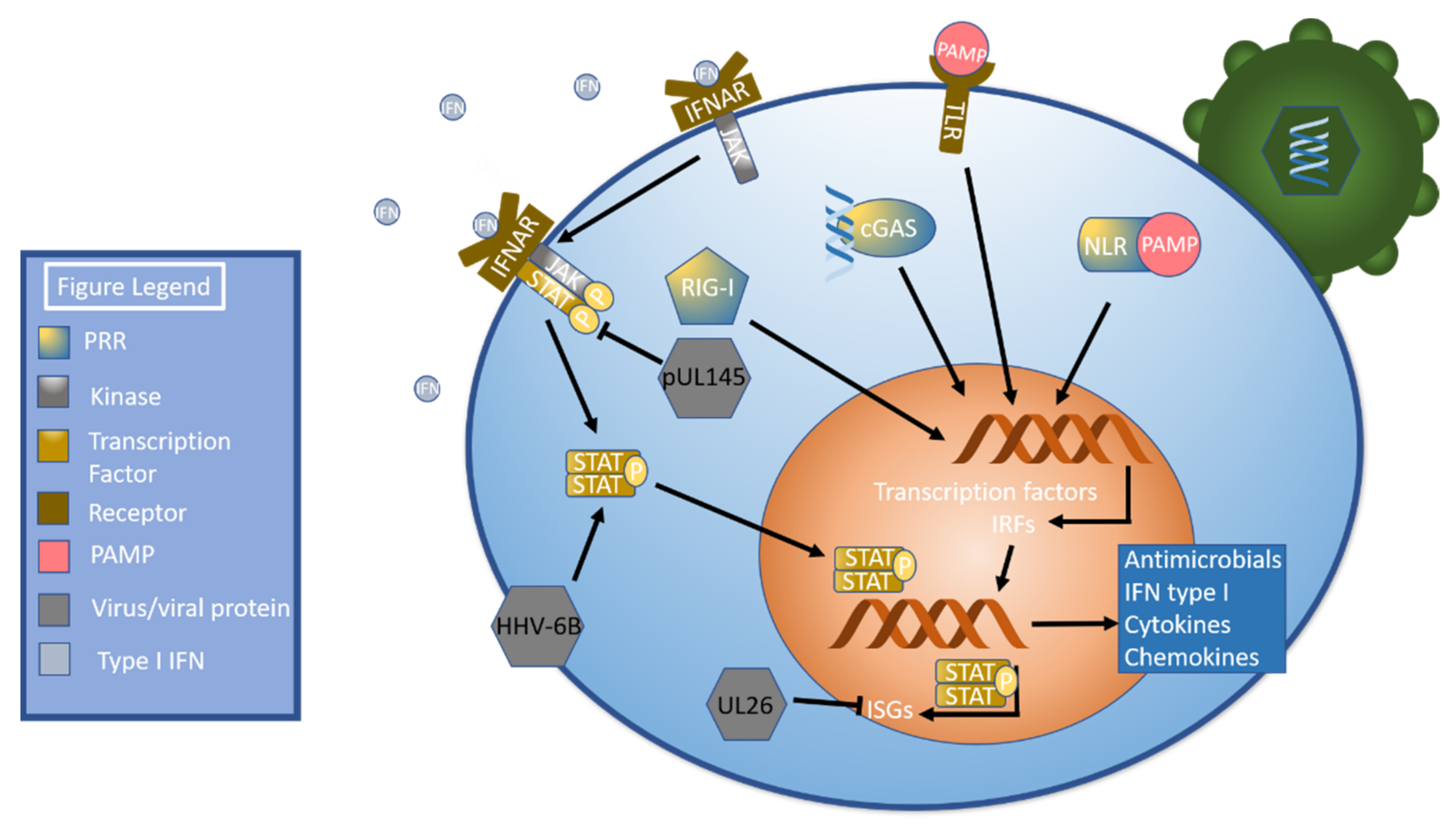
3. Immune Cell Responses
4. Autophagy and Apoptosis
|
Virus |
Immunoevasive Strategy |
Associated References |
|---|---|---|
|
HCMV |
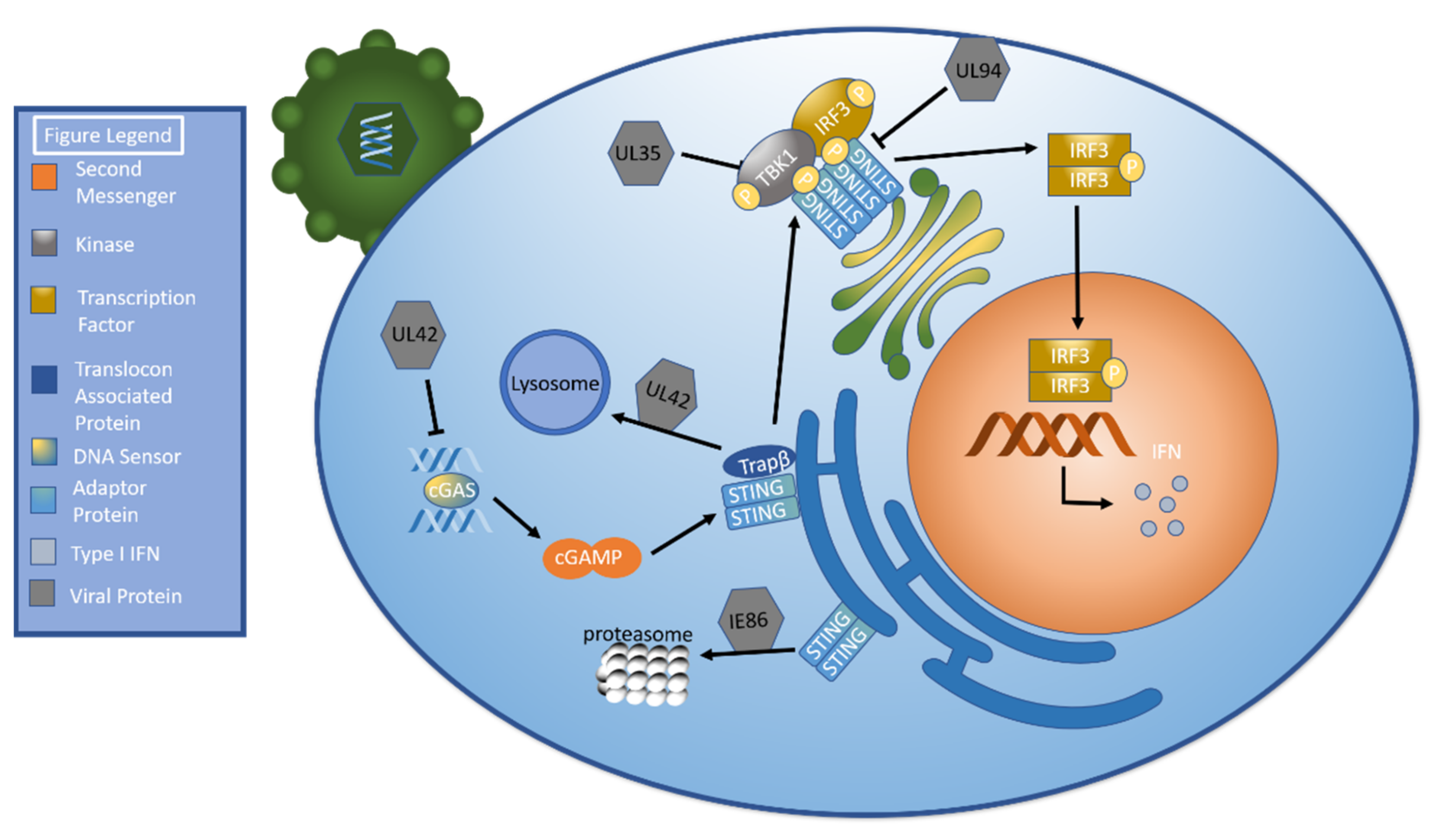
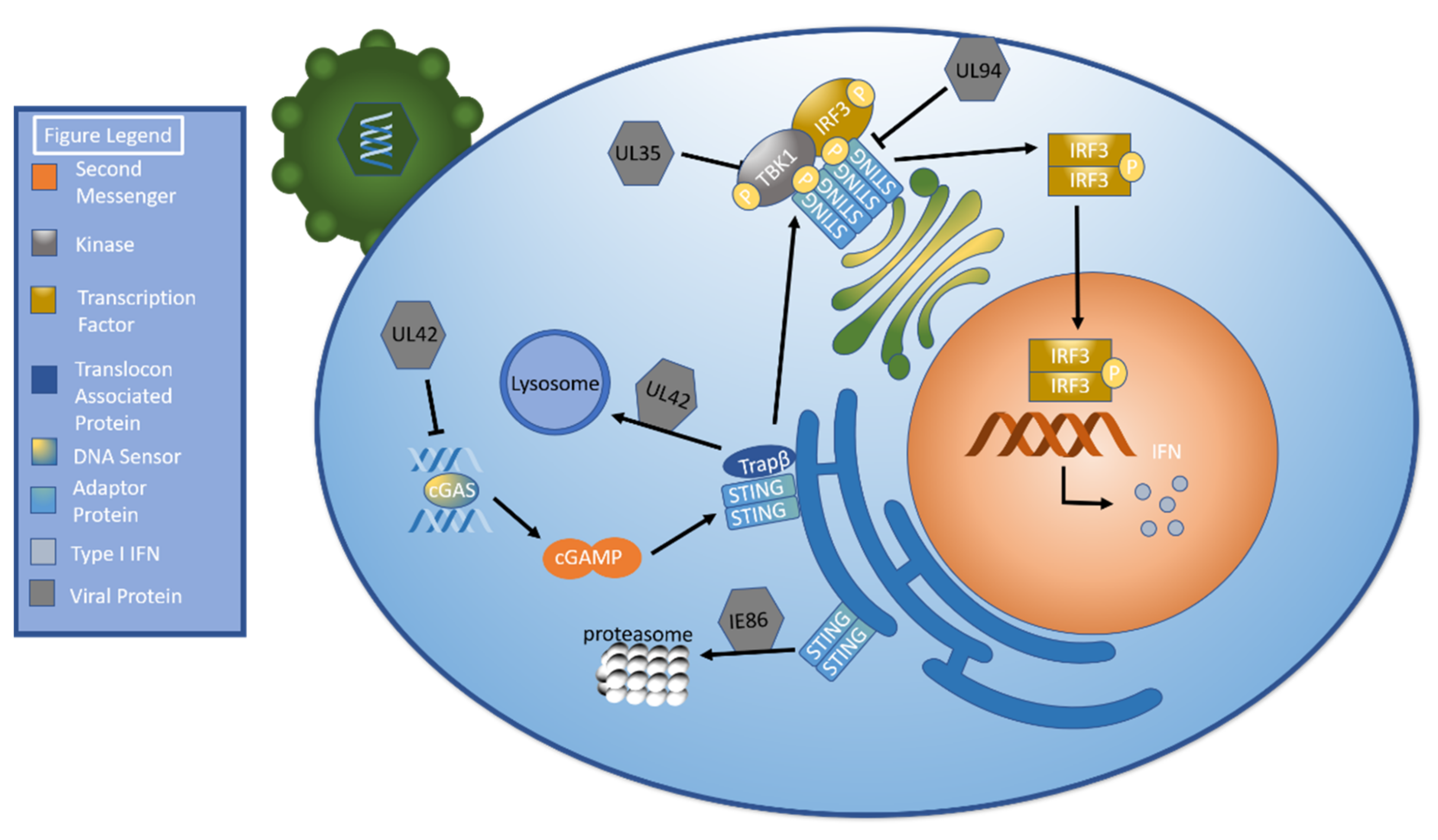
UL35: Decrease TBK/IRF3 phosphorylation |
[70] |
|
UL42: Inhibit cGAS’ ability to bind HCMV DNA Inhibit cGAS oligomerization Facilitate TRAPβ degradation (lysosome) |
[71] |
|
|
UL94: Inhibit STING dimerization Prevent STING from recruiting TBK1 |
[72] |
|
|
IE86: Facilitate STING degradation via proteasome |
||
|
US7: Facilitate TLR3 and TLR4 proteasomal degradation |
[75] |
|
|
US8: Inhibit TLR3 and TLR4 by destabilization |
[75] |
|
|
IE1: Suppress CpG motifs to avoid ZAP detection |
[76] |
|
|
UL26: Inhibit ISG15/ISGylation |
[77] |
|
|
pUL145: Downregulate STAT |
[78] |
|
|
UL147a: Target MICA for lysosomal degradation |
[79] |
|
|
UL148a: Target MICA for lysosomal degradation |
[80] |
|
|
gB: Interact with epidermal growth factor receptor to stimulate the AKT pathway |
||
|
gH: Interact with αvβ3 integrin to stimulate the AKT pathway |
||
|
pUL7: Stimulate phosphorylation, translocation to cytoplasm, and inactivation of FOXO3a |
[64] |
|
|
miR-US5-1 and miR-UL112-3p: FOXO3a inhibition |
[64] |
|
|
Downregulate CIITA |
[81] |
|
|
Downregulate MHC class I expression |
[82] |
|
|
Alter expression of apoptotic genes |
[52] |
|
|
HHV-6A |
Downregulate IFI16 |
[83] |
|
Induce phosphorylation of STAT6 |
[83] |
|
|
Inhibit TLR9 mRNA and protein expression |
[83] |
|
|
Infect NK cells and alter miRNA and transcription factor expression |
[24] |
|
|
Modulate HLA class I and class II expression |
[84] |
|
|
Alter T lymphocyte miRNA upon infection |
[85] |
|
|
Favor TH2 cytokine profile upon dendritic cell infection |
[86] |
|
|
Alter expression of apoptotic genes |
[52] |
|
|
Increase autophagic flux |
[67] |
|
|
Increase BiP |
[67] |
|
|
HHV-6B |
Downregulate IFI16 |
[83] |
|
Inhibit TLR9 protein levels |
[83] |
|
|
Inhibit TLR signaling |
[87] |
|
|
Inhibit autophagy |
||
|
Upregulate CHOP via modulating expression of IRE1α, ATF4, and ATF6 |
[67] |
|
|
Inhibit monocyte survival and differentiation into dendritic cells |
[69] |
|
|
Increase ROS to upregulate PD-L1, influence STAT phosphorylation |
[88] |
|
|
Downregulate NKG2D ligands |
[89] |
|
|
Infect NK cells and alter miRNA and transcription factor expression |
[24] |
|
|
HHV-7 |
U21: Targets MCH class I complex for lysosomal degradation, target UBPL1 for lysosomal degradiation, decrease MICA and MICB concentration, post-transcriptional modification of MICA and MICB |
|
|
Slight decrease in cGAS expression |
[83] |
5. Latency and Reactivation
6. Vaccination Efforts
References
- Cohen, J.I. Herpesvirus latency. J. Clin. Invest. 2020, 130, 3361–3369.
- Ablashi, D.V.; Berneman, Z.N.; Kramarsky, B.; Asano, Y.; Choudhury, S.; Pearson, G.R. Human herpesvirus-7 (HHV-7). Vivo 1994, 8, 549–554.
- Campadelli-Fiume, G.; Mirandola, P.; Menotti, L. Human herpesvirus 6: An emerging pathogen. Emerg. Infect. Dis. 1999, 5, 353–366.
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–1822.
- Close, W.L.; Anderson, A.N.; Pellett, P.E. Betaherpesvirus Virion Assembly and Egress. Adv. Exp. Med. Biol. 2018, 1045, 167–207.
- Tomtishen, J.P., 3rd. Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28). Virol. J. 2012, 9, 22.
- Full, F.; Ensser, A. Early Nuclear Events after Herpesviral Infection. J. Clin. Med. 2019, 8, 1408.
- Guo, H.; Shen, S.; Wang, L.; Deng, H. Role of tegument proteins in herpesvirus assembly and egress. Protein Cell 2010, 1, 987–998.
- Nishimura, M.; Novita, B.D.; Kato, T.; Handayani Tjan, L.; Wang, B.; Wakata, A.; Lystia Poetranto, A.; Kawabata, A.; Tang, H.; Aoshi, T.; et al. Structural basis for the interaction of human herpesvirus 6B tetrameric glycoprotein complex with the cellular receptor, human CD134. PLoS Pathog. 2020, 16, e1008648.
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121.
- Gugliesi, F.; Coscia, A.; Griffante, G.; Galitska, G.; Pasquero, S.; Albano, C.; Biolatti, M. Where do we Stand after Decades of Studying Human Cytomegalovirus? Microorganisms 2020, 8, 685.
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704.
- Gerna, G.; Kabanova, A.; Lilleri, D. Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines 2019, 7, 70.
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Laboratory and clinical aspects of human herpesvirus 6 infections. Clin. Microbiol. Rev. 2015, 28, 313–335.
- Mori, Y. Recent topics related to human herpesvirus 6 cell tropism. Cell Microbiol. 2009, 11, 1001–1006.
- Dockrell, D.H. Human herpesvirus 6: Molecular biology and clinical features. J. Med. Microbiol. 2003, 52, 5–18.
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Update on infections with human herpesviruses 6A, 6B, and 7. Med. Mal. Infect. 2017, 47, 83–91.
- Scrivano, L.; Sinzger, C.; Nitschko, H.; Koszinowski, U.H.; Adler, B. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog. 2011, 7, e1001256.
- Landolfo, S.; Gariglio, M.; Gribaudo, G.; Lembo, D. The human cytomegalovirus. Pharmacol. Ther. 2003, 98, 269–297.
- Dutt, K.; Ezeonu, I. Human retinal and brain cell lines: A model of HCMV retinitis and encephalitis. DNA Cell Biol. 2006, 25, 581–596.
- Minsart, A.F.; Rypens, F.; Smiljkovic, M.; Kakkar, F.; Renaud, C.; Lamarre, V.; Boucher, M.; Boucoiran, I. Prenatal findings, neonatal symptoms and neurodevelopmental outcome of congenital cytomegalovirus infection in a university hospital in Montreal, Quebec. J. Perinat. Med. 2020, 48, 234–241.
- Cohen, J.I.; Corey, G.R. Cytomegalovirus infection in the normal host. Medicine 1985, 64, 100–114.
- Tang, H.; Mori, Y. Glycoproteins of HHV-6A and HHV-6B. Adv. Exp. Med. Biol. 2018, 1045, 145–165.
- Rizzo, R.; Soffritti, I.; D’Accolti, M.; Bortolotti, D.; Di Luca, D.; Caselli, E. HHV-6A/6B Infection of NK Cells Modulates the Expression of miRNAs and Transcription Factors Potentially Associated to Impaired NK Activity. Front. Microbiol. 2017, 8, 2143.
- Ablashi, D.; Agut, H.; Alvarez-Lafuente, R.; Clark, D.A.; Dewhurst, S.; DiLuca, D.; Flamand, L.; Frenkel, N.; Gallo, R.; Gompels, U.A.; et al. Classification of HHV-6A and HHV-6B as distinct viruses. Arch. Virol. 2014, 159, 863–870.
- Marci, R.; Gentili, V.; Bortolotti, D.; Lo Monte, G.; Caselli, E.; Bolzani, S.; Rotola, A.; Di Luca, D.; Rizzo, R. Presence of HHV-6A in Endometrial Epithelial Cells from Women with Primary Unexplained Infertility. PLoS ONE 2016, 11, e0158304.
- Hansen, A.S.; Bundgaard, B.B.; Biltoft, M.; Rossen, L.S.; Hollsberg, P. Divergent tropism of HHV-6AGS and HHV-6BPL1 in T cells expressing different CD46 isoform patterns. Virology 2017, 502, 160–170.
- Ongradi, J.; Ablashi, D.V.; Yoshikawa, T.; Stercz, B.; Ogata, M. Roseolovirus-associated encephalitis in immunocompetent and immunocompromised individuals. J. Neurovirol. 2017, 23, 1–19.
- Hill, J.A. Human herpesvirus 6 in transplant recipients: An update on diagnostic and treatment strategies. Curr. Opin. Infect. Dis. 2019, 32, 584–590.
- Leibovitch, E.C.; Jacobson, S. Evidence linking HHV-6 with multiple sclerosis: An update. Curr. Opin. Virol. 2014, 9, 127–133.
- Soldan, S.S.; Berti, R.; Salem, N.; Secchiero, P.; Flamand, L.; Calabresi, P.A.; Brennan, M.B.; Maloni, H.W.; McFarland, H.F.; Lin, H.C.; et al. Association of human herpes virus 6 (HHV-6) with multiple sclerosis: Increased IgM response to HHV-6 early antigen and detection of serum HHV-6 DNA. Nat. Med. 1997, 3, 1394–1397.
- Itzhaki, R.F.; Golde, T.E.; Heneka, M.T.; Readhead, B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat. Rev. Neurol. 2020, 16, 193–197.
- Tarlinton, R.E.; Martynova, E.; Rizvanov, A.A.; Khaiboullina, S.; Verma, S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses 2020, 12, 643.
- Friedman, J.E.; Lyons, M.J.; Cu, G.; Ablashl, D.V.; Whitman, J.E.; Edgar, M.; Koskiniemi, M.; Vaheri, A.; Zabriskie, J.B. The association of the human herpesvirus-6 and MS. Mult. Scler. 1999, 5, 355–362.
- Zabalza, A.; Vera, A.; Alari-Pahissa, E.; Munteis, E.; Moreira, A.; Yelamos, J.; Llop, M.; Lopez-Botet, M.; Martinez-Rodriguez, J.E. Impact of cytomegalovirus infection on B cell differentiation and cytokine production in multiple sclerosis. J. Neuroinflammation 2020, 17, 161.
- Nora-Krukle, Z.; Chapenko, S.; Logina, I.; Millers, A.; Platkajis, A.; Murovska, M. Human herpesvirus 6 and 7 reactivation and disease activity in multiple sclerosis. Medicina 2011, 47, 527–531.
- Sepulveda, N.; Carneiro, J.; Lacerda, E.; Nacul, L. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome as a Hyper-Regulated Immune System Driven by an Interplay Between Regulatory T Cells and Chronic Human Herpesvirus Infections. Front. Immunol. 2019, 10, 2684.
- Chapenko, S.; Krumina, A.; Kozireva, S.; Nora, Z.; Sultanova, A.; Viksna, L.; Murovska, M. Activation of human herpesviruses 6 and 7 in patients with chronic fatigue syndrome. J. Clin. Virol. 2006, 37 (Suppl. 1), S47–S51.
- Simpson, S., Jr.; Taylor, B.; Burrows, J.; Burrows, S.; Dwyer, D.E.; Taylor, J.; Ponsonby, A.L.; Blizzard, L.; Dwyer, T.; Pittas, F.; et al. EBV & HHV6 reactivation is infrequent and not associated with MS clinical course. Acta Neurol. Scand. 2014, 130, 328–337.
- Soto, N.E.; Straus, S.E. Chronic Fatigue Syndrome and Herpesviruses: The Fading Evidence. Herpes 2000, 7, 46–50.
- Buchwald, D.; Ashley, R.L.; Pearlman, T.; Kith, P.; Komaroff, A.L. Viral serologies in patients with chronic fatigue and chronic fatigue syndrome. J. Med. Virol. 1996, 50, 25–30.
- Kumar, S.; Ingle, H.; Prasad, D.V.; Kumar, H. Recognition of bacterial infection by innate immune sensors. Crit. Rev. Microbiol. 2013, 39, 229–246.
- Paludan, S.R.; Pradeu, T.; Masters, S.L.; Mogensen, T.H. Constitutive immune mechanisms: Mediators of host defence and immune regulation. Nat. Rev. Immunol. 2021, 21, 137–150.
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol 2014, 14, 36–49.
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629.
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353.
- Thery, C.; Amigorena, S. The cell biology of antigen presentation in dendritic cells. Curr. Opin. Immunol. 2001, 13, 45–51.
- Hammer, Q.; Romagnani, C. About Training and Memory: NK-Cell Adaptation to Viral Infections. Adv. Immunol. 2017, 133, 171–207.
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120.
- Lee, E.W.; Seo, J.; Jeong, M.; Lee, S.; Song, J. The roles of FADD in extrinsic apoptosis and necroptosis. BMB Rep. 2012, 45, 496–508.
- Orzalli, M.H.; Kagan, J.C. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol. 2017, 27, 800–809.
- Arcangeletti, M.C.; D’Accolti, M.; Maccari, C.; Soffritti, I.; Conto, F.; Chezzi, C.; Calderaro, A.; Ferri, C.; Caselli, E. Impact of Human Cytomegalovirus and Human Herpesvirus 6 Infection on the Expression of Factors Associated with Cell Fibrosis and Apoptosis: Clues for Implication in Systemic Sclerosis Development. Int. J. Mol. Sci. 2020, 21, 6397.
- Smith, M.S.; Bentz, G.L.; Alexander, J.S.; Yurochko, A.D. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J. Virol. 2004, 78, 4444–4453.
- Van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435.
- Goyal, A.; Wang, Y.; Graham, M.M.; Doseff, A.I.; Bhatt, N.Y.; Marsh, C.B. Monocyte survival factors induce Akt activation and suppress caspase-3. Am. J. Respir Cell Mol. Biol. 2002, 26, 224–230.
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555.
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450.
- Wan, H.; Xie, T.; Xu, Q.; Hu, X.; Xing, S.; Yang, H.; Gao, Y.; He, Z. Thy-1 depletion and integrin beta3 upregulation-mediated PI3K-Akt-mTOR pathway activation inhibits lung fibroblast autophagy in lipopolysaccharide-induced pulmonary fibrosis. Lab. Invest. 2019, 99, 1636–1649.
- Cojohari, O.; Peppenelli, M.A.; Chan, G.C. Human Cytomegalovirus Induces an Atypical Activation of Akt To Stimulate the Survival of Short-Lived Monocytes. J. Virol. 2016, 90, 6443–6452.
- Peppenelli, M.A.; Arend, K.C.; Cojohari, O.; Moorman, N.J.; Chan, G.C. Human Cytomegalovirus Stimulates the Synthesis of Select Akt-Dependent Antiapoptotic Proteins during Viral Entry To Promote Survival of Infected Monocytes. J. Virol. 2016, 90, 3138–3147.
- Mahmud, J.; Miller, M.J.; Altman, A.M.; Chan, G.C. Human Cytomegalovirus Glycoprotein-Initiated Signaling Mediates the Aberrant Activation of Akt. J. Virol. 2020, 94.
- Kerr, W.G. Inhibitor and activator: Dual functions for SHIP in immunity and cancer. Ann. N. Y. Acad. Sci. 2011, 1217, 1–17.
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986.
- Hancock, M.H.; Crawford, L.B.; Perez, W.; Struthers, H.M.; Mitchell, J.; Caposio, P. Human Cytomegalovirus UL7, miR-US5-1, and miR-UL112-3p Inactivation of FOXO3a Protects CD34(+) Hematopoietic Progenitor Cells from Apoptosis. mSphere 2021, 6.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741.
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Munz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024.
- Romeo, M.A.; Masuelli, L.; Gaeta, A.; Nazzari, C.; Granato, M.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Impact of HHV-6A and HHV-6B lytic infection on autophagy and endoplasmic reticulum stress. J. Gen. Virol. 2019, 100, 89–98.
- Qi, Z.; Chen, L. Endoplasmic Reticulum Stress and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 167–177.
- Romeo, M.A.; Gilardini Montani, M.S.; Falcinelli, L.; Gaeta, A.; Nazzari, C.; Faggioni, A.; Cirone, M. HHV-6B reduces autophagy and induces ER stress in primary monocytes impairing their survival and differentiation into dendritic cells. Virus Res. 2019, 273, 197757.
- Fabits, M.; Goncalves Magalhaes, V.; Chan, B.; Girault, V.; Elbasani, E.; Rossetti, E.; Saeland, E.; Messerle, M.; Pichlmair, A.; Lisnic, V.J.; et al. The Cytomegalovirus Tegument Protein UL35 Antagonizes Pattern Recognition Receptor-Mediated Type I IFN Transcription. Microorganisms 2020, 8, 790.
- Fu, Y.Z.; Guo, Y.; Zou, H.M.; Su, S.; Wang, S.Y.; Yang, Q.; Luo, M.H.; Wang, Y.Y. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS Pathog. 2019, 15, e1007691.
- Zou, H.M.; Huang, Z.F.; Yang, Y.; Luo, W.W.; Wang, S.Y.; Luo, M.H.; Fu, Y.Z.; Wang, Y.Y. Human Cytomegalovirus Protein UL94 Targets MITA to Evade the Antiviral Immune Response. J. Virol. 2020, 94.
- Kim, J.E.; Kim, Y.E.; Stinski, M.F.; Ahn, J.H.; Song, Y.J. Human Cytomegalovirus IE2 86 kDa Protein Induces STING Degradation and Inhibits cGAMP-Mediated IFN-beta Induction. Front. Microbiol. 2017, 8, 1854.
- Lee, J.K.; Kim, J.E.; Park, B.J.; Song, Y.J. Human cytomegalovirus IE86 protein aa 136-289 mediates STING degradation and blocks the cGAS-STING pathway. J. Microbiol. 2020, 58, 54–60.
- Park, A.; Ra, E.A.; Lee, T.A.; Choi, H.J.; Lee, E.; Kang, S.; Seo, J.Y.; Lee, S.; Park, B. HCMV-encoded US7 and US8 act as antagonists of innate immunity by distinctively targeting TLR-signaling pathways. Nat. Commun. 2019, 10, 4670.
- Lin, Y.T.; Chiweshe, S.; McCormick, D.; Raper, A.; Wickenhagen, A.; DeFillipis, V.; Gaunt, E.; Simmonds, P.; Wilson, S.J.; Grey, F. Human cytomegalovirus evades ZAP detection by suppressing CpG dinucleotides in the major immediate early 1 gene. PLoS Pathog. 2020, 16, e1008844.
- Goodwin, C.M.; Schafer, X.; Munger, J. UL26 Attenuates IKKbeta-Mediated Induction of Interferon-Stimulated Gene (ISG) Expression and Enhanced Protein ISGylation during Human Cytomegalovirus Infection. J. Virol. 2019, 93.
- Le-Trilling, V.T.K.; Becker, T.; Nachshon, A.; Stern-Ginossar, N.; Scholer, L.; Voigt, S.; Hengel, H.; Trilling, M. The Human Cytomegalovirus pUL145 Isoforms Act as Viral DDB1-Cullin-Associated Factors to Instruct Host Protein Degradation to Impede Innate Immunity. Cell Rep. 2020, 30, 2248–2260 e2245.
- Seidel, E.; Dassa, L.; Schuler, C.; Oiknine-Djian, E.; Wolf, D.G.; Le-Trilling, V.T.K.; Mandelboim, O. The human cytomegalovirus protein UL147A downregulates the most prevalent MICA allele: MICA*008, to evade NK cell-mediated killing. PLoS Pathog. 2021, 17, e1008807.
- Dassa, L.; Seidel, E.; Oiknine-Djian, E.; Yamin, R.; Wolf, D.G.; Le-Trilling, V.T.K.; Mandelboim, O. The Human Cytomegalovirus Protein UL148A Downregulates the NK Cell-Activating Ligand MICA to Avoid NK Cell Attack. J. Virol. 2018, 92.
- Sandhu, P.K.; Buchkovich, N.J. Human Cytomegalovirus Decreases Major Histocompatibility Complex Class II by Regulating Class II Transactivator Transcript Levels in a Myeloid Cell Line. J. Virol. 2020, 94.
- Abdelaziz, M.O.; Ossmann, S.; Kaufmann, A.M.; Leitner, J.; Steinberger, P.; Willimsky, G.; Raftery, M.J.; Schonrich, G. Development of a Human Cytomegalovirus (HCMV)-Based Therapeutic Cancer Vaccine Uncovers a Previously Unsuspected Viral Block of MHC Class I Antigen Presentation. Front. Immunol. 2019, 10, 1776.
- Bortolotti, D.; Gentili, V.; Caselli, E.; Sicolo, M.; Soffritti, I.; D’Accolti, M.; Barao, I.; Rotola, A.; Di Luca, D.; Rizzo, R. DNA Sensors’ Signaling in NK Cells During HHV-6A, HHV-6B and HHV-7 Infection. Front. Microbiol. 2020, 11, 226.
- Caselli, E.; Campioni, D.; Cavazzini, F.; Gentili, V.; Bortolotti, D.; Cuneo, A.; Di Luca, D.; Rizzo, R. Acute human herpesvirus-6A infection of human mesothelial cells modulates HLA molecules. Arch. Virol. 2015, 160, 2141–2149.
- Caselli, E.; D’Accolti, M.; Soffritti, I.; Zatelli, M.C.; Rossi, R.; Degli Uberti, E.; Di Luca, D. HHV-6A in vitro infection of thyrocytes and T cells alters the expression of miRNA associated to autoimmune thyroiditis. Virol. J. 2017, 14, 3.
- Gustafsson, R. Human Herpesvirus 6A Induces Dendritic Cell Death and HMGB1 Release without Virus Replication. Pathogens 2021, 10, 57.
- Murakami, Y.; Tanimoto, K.; Fujiwara, H.; An, J.; Suemori, K.; Ochi, T.; Hasegawa, H.; Yasukawa, M. Human herpesvirus 6 infection impairs Toll-like receptor signaling. Virol. J. 2010, 7, 91.
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Giambelli, L.; D’Aprile, R.; Gaeta, A.; Faggioni, A.; Cirone, M. The cross-talk between STAT1/STAT3 and ROS up-regulates PD-L1 and promotes the release of pro-inflammatory/immune suppressive cytokines in primary monocytes infected by HHV-6B. Virus Res. 2021, 292, 198231.
- Schmiedel, D.; Tai, J.; Levi-Schaffer, F.; Dovrat, S.; Mandelboim, O. Human Herpesvirus 6B Downregulates Expression of Activating Ligands during Lytic Infection To Escape Elimination by Natural Killer Cells. J. Virol. 2016, 90, 9608–9617.
- May, N.A.; Glosson, N.L.; Hudson, A.W. Human herpesvirus 7 u21 downregulates classical and nonclassical class I major histocompatibility complex molecules from the cell surface. J. Virol. 2010, 84, 3738–3751.
- Hudson, A.W.; Howley, P.M.; Ploegh, H.L. A human herpesvirus 7 glycoprotein, U21, diverts major histocompatibility complex class I molecules to lysosomes. J. Virol. 2001, 75, 12347–12358.
- Schneider, C.L.; Hudson, A.W. The human herpesvirus-7 (HHV-7) U21 immunoevasin subverts NK-mediated cytoxicity through modulation of MICA and MICB. PLoS Pathog 2011, 7, e1002362.
- Dirck, A.T.; Whyte, M.L.; Hudson, A.W. HHV-7 U21 exploits Golgi quality control carriers to reroute class I MHC molecules to lysosomes. Mol. Biol. Cell 2020, 31, 196–208.
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 2013, 4, e00332-13.
- Liu, J.; Jardetzky, T.S.; Chin, A.L.; Johnson, D.C.; Vanarsdall, A.L. The Human Cytomegalovirus Trimer and Pentamer Promote Sequential Steps in Entry into Epithelial and Endothelial Cells at Cell Surfaces and Endosomes. J. Virol. 2018, 92.
- Tang, H.; Wang, J.; Mahmoud, N.F.; Mori, Y. Detailed study of the interaction between human herpesvirus 6B glycoprotein complex and its cellular receptor, human CD134. J. Virol. 2014, 88, 10875–10882.
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutre, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect. Dis. 2017, 17, e177–e188.
- Legendre, C.; Pascual, M. Improving outcomes for solid-organ transplant recipients at risk from cytomegalovirus infection: Late-onset disease and indirect consequences. Clin. Infect. Dis 2008, 46, 732–740.
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297.
- Nelson, C.S.; Jenks, J.A.; Pardi, N.; Goodwin, M.; Roark, H.; Edwards, W.; McLellan, J.S.; Pollara, J.; Weissman, D.; Permar, S.R. Human Cytomegalovirus Glycoprotein B Nucleoside-Modified mRNA Vaccine Elicits Antibody Responses with Greater Durability and Breadth than MF59-Adjuvanted gB Protein Immunization. J. Virol. 2020, 94.
- Perotti, M.; Marcandalli, J.; Demurtas, D.; Sallusto, F.; Perez, L. Rationally designed Human Cytomegalovirus gB nanoparticle vaccine with improved immunogenicity. PLoS Pathog. 2020, 16, e1009169.
- Lopez-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-assembling protein nanoparticles in the design of vaccines. Comput. Struct. Biotechnol. J. 2016, 14, 58–68.
- Kern, F.; Bunde, T.; Faulhaber, N.; Kiecker, F.; Khatamzas, E.; Rudawski, I.M.; Pruss, A.; Gratama, J.W.; Volkmer-Engert, R.; Ewert, R.; et al. Cytomegalovirus (CMV) phosphoprotein 65 makes a large contribution to shaping the T cell repertoire in CMV-exposed individuals. J. Infect. Dis. 2002, 185, 1709–1716.
- Chiuppesi, F.; Nguyen, J.; Park, S.; Contreras, H.; Kha, M.; Meng, Z.; Kaltcheva, T.; Iniguez, A.; Martinez, J.; La Rosa, C.; et al. Multiantigenic Modified Vaccinia Virus Ankara Vaccine Vectors To Elicit Potent Humoral and Cellular Immune Reponses against Human Cytomegalovirus in Mice. J. Virol. 2018, 92.
- Choi, K.Y.; El-Hamdi, N.S.; McGregor, A. Inclusion of the Viral Pentamer Complex in a Vaccine Design Greatly Improves Protection against Congenital Cytomegalovirus in the Guinea Pig Model. J. Virol. 2019, 93.
- Coleman, S.; Hornig, J.; Maddux, S.; Choi, K.Y.; McGregor, A. Viral Glycoprotein Complex Formation, Essential Function and Immunogenicity in the Guinea Pig Model for Cytomegalovirus. PLoS ONE 2015, 10, e0135567.
- Roark, H.K.; Jenks, J.A.; Permar, S.R.; Schleiss, M.R. Animal Models of Congenital Cytomegalovirus Transmission: Implications for Vaccine Development. J. Infect. Dis. 2020, 221, S60–S73.
- Plotkin, S.A.; Wang, D.; Oualim, A.; Diamond, D.J.; Kotton, C.N.; Mossman, S.; Carfi, A.; Anderson, D.; Dormitzer, P.R. The Status of Vaccine Development against the Human Cytomegalovirus. J. Infect. Dis. 2020, 221, S113–S122.
- Inoue, N.; Abe, M.; Kobayashi, R.; Yamada, S. Vaccine Development for Cytomegalovirus. Adv. Exp. Med. Biol. 2018, 1045, 271–296.
- Plotkin, S.A. Preventing Infection by Human Cytomegalovirus. J. Infect. Dis. 2020, 221, S123–S127.
- Akkapaiboon, P.; Mori, Y.; Sadaoka, T.; Yonemoto, S.; Yamanishi, K. Intracellular processing of human herpesvirus 6 glycoproteins Q1 and Q2 into tetrameric complexes expressed on the viral envelope. J. Virol. 2004, 78, 7969–7983.
- Tang, H.; Hayashi, M.; Maeki, T.; Yamanishi, K.; Mori, Y. Human herpesvirus 6 glycoprotein complex formation is required for folding and trafficking of the gH/gL/gQ1/gQ2 complex and its cellular receptor binding. J. Virol. 2011, 85, 11121–11130.
- Tang, H.; Serada, S.; Kawabata, A.; Ota, M.; Hayashi, E.; Naka, T.; Yamanishi, K.; Mori, Y. CD134 is a cellular receptor specific for human herpesvirus-6B entry. Proc. Natl. Acad. Sci. USA 2013, 110, 9096–9099.
- Mori, Y.; Seya, T.; Huang, H.L.; Akkapaiboon, P.; Dhepakson, P.; Yamanishi, K. Human herpesvirus 6 variant A but not variant B induces fusion from without in a variety of human cells through a human herpesvirus 6 entry receptor, CD46. J. Virol. 2002, 76, 6750–6761.
- Wang, B.; Hara, K.; Kawabata, A.; Nishimura, M.; Wakata, A.; Tjan, L.H.; Poetranto, A.L.; Yamamoto, C.; Haseda, Y.; Aoshi, T.; et al. Tetrameric glycoprotein complex gH/gL/gQ1/gQ2 is a promising vaccine candidate for human herpesvirus 6B. PLoS Pathog. 2020, 16, e1008609.

