The human immune system boasts a diverse array of strategies for recognizing and eradicating invading pathogens. Human betaherpesviruses, a highly prevalent subfamily of viruses, include human cytomegalovirus (HCMV), human herpesvirus (HHV) 6A, HHV-6B, and HHV-7. These viruses have evolved numerous mechanisms for evading the host response.
1. Introduction
Betaherpesviruses, a widespread subfamily of viruses within the herpesviridae family, are nearly ubiquitous in the global population [
1,
2,
3] and include human cytomegalovirus (HCMV), human herpesvirus (HHV) 6A, HHV-6B, and HHV-7 [
4].
Like other herpesviridae, betaherpesviruses are comprised of four primary sections: the double-stranded viral DNA, the capsid, the tegument, and the envelope. The capsid surrounds the DNA core, while the tegument is a protein-filled area surrounding the capsid. The envelope, the most exterior structural component of herpesviridae, is studded with glycoprotein spikes [
4,
5]. Tegument proteins are involved in a wide variety of viral activities, including virion assembly, gene expression, egress, and immune evasion [
6,
7,
8]. Envelope glycoproteins play a particularly prominent role in protection and in mediating viral entry [
5,
9,
10].
The tropism of HCMV [
11,
12,
13], HHV-6 [
14,
15,
16,
17], and HHV-7 [
17] has been reviewed in depth elsewhere. Specific cellular receptors and viral proteins play an important role in the determination of viral tropism and are also detailed in the aforementioned reviews. HCMV can infect a variety of different cell types in vivo and in vitro, including epithelial cells, endothelial cells, fibroblasts, and smooth muscle cells [
18]. While many receptors contribute to viral entry in HCMV, there are two models of viral entry, one for fibroblasts and one for epithelial and endothelial cells [
13]. In fibroblasts, the cellular platelet-derived growth factor-α receptor (PDGFRα) interacts with the HCMV gH/gL/gO trimer complex (TC), which activates glycoprotein B (gB) to fuse the viral envelope with the cell membrane [
13]. In epithelial and endothelial cells, the cellular receptor neuropilin2 (Nrp2) interacts with the HCMV gH/gL/pUL128L pentamer complex (PC) to stimulate viral endocytosis followed by TC activation of gB, which facilitates viral entry [
13]. Once viral entry is accomplished, local spread is mediated by direct cell-to-cell transmission [
13]. However, HCMV primarily establishes latency in cells arising from myeloid progenitors. HCMV has been found to infect dendritic cells latently as well as peripheral mononuclear blood cells, including monocytes and CD34+ progenitor cells [
11]. Infection begins following exposure to contaminated saliva, mucosa, skin lesions, or genital secretions [
1]. HCMV disease associations include transplant rejection, retinitis, and serious birth defects [
19,
20,
21]. HCMV can also present as a mono-like illness [
22].
HHV-6 also infects a broad range of cells in vivo and in vitro, although it preferentially infects activated CD4+ T lymphocytes. In vivo, HHV-6 can infect tissues ranging from the brain to the liver [
14] as well as a variety of additional cell types including endothelial cells, NK cells, and myeloid cells [
23,
24]. HHV 6 is composed of two related but distinct viruses, HHV-6A and HHV-6B [
25]. HHV-6A but not HHV-6B has been shown to infect endometrial cells [
26]. However, HHV-6A and HHV-6B differ in their replicative ability within specific transformed T-lymphocyte cell lines [
15,
16]. For example, HHV-6A is most commonly maintained in HSB-2 or JJhan T cell lines while HHV-6B is preferentially maintained in Molt3 or MT4 [
15,
23]. In addition, a recent study showed that HHV-6A
GS was capable of transcribing the viral genes U7 and U23 in the T cell lines Peer and Jurkat while HHV-6B
PL1 could not. Both HHV-6A and HHV-6B infect Molt3 and SupT1 T cells [
27]. HHV-6A uses the cell surface receptor CD46 to mediate viral entry, while HHV-6B uses CD134 [
14,
17]. HHV-7 has similar tropism to HHV-6, selectively infecting CD4+ T cells. However, HHV-7 can be observed in various tissues in vivo, including the skin, salivary glands, and other organs [
17]. Like HCMV, contaminated saliva, mucosa, skin lesions, or genital secretions leads to HHV-6 and HHV-7 infection [
1]. The primary disease associated with HHV-6 is exanthem subitum, commonly known as roseola or sixth disease, a self-limited disease seen in infants aged 6 months to 2 years of age [
14]. More serious disease associations include seizures, encephalitis/encephalopathy, retinitis, and hepatitis [
14,
28]. Complications including graft versus host disease and lower respiratory tract infections have been reported in the setting of organ transplantation [
29]. HHV-6 has also been associated with neurodegenerative disorders such as multiple sclerosis [
30,
31] and Alzheimer’s disease [
32], although a definitive link has yet to be established. HHV-7 is a sparsely researched virus with limited disease associations at this time, although it is also thought to cause exanthem subitum [
17]. It, too, has been associated with encephalitis/encephalopathy [
28] and more tenuously with Alzheimer’s disease [
32]. Betaherpesviruses have also been associated with multiple sclerosis [
33,
34,
35,
36] and chronic fatigue syndrome [
37,
38], although these associations have been questioned in the past [
39,
40,
41].
2. Innate Immune Response Pathways
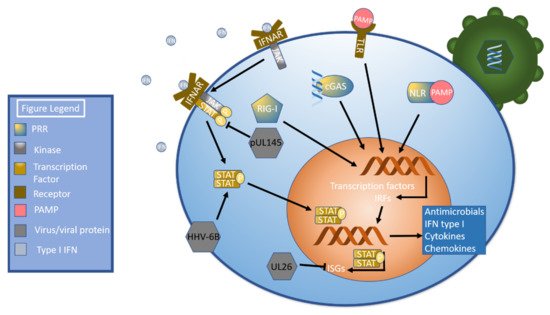
A primary mechanism to induce the innate immune response begins when pathogen associated molecular patterns (PAMPs) are detected by pattern recognition receptors (PRRs), including toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors, and cGAS-STING [
42,
43]. Pattern recognition receptor activation stimulates signal transduction pathways that culminate in the activation of transcription factors including NF-κB and interferon regulatory factors. These transcription factors drive the production of antimicrobial peptides, proinflammatory cytokines, chemokines, and type I interferons [
42]. Type I Interferon (IFN) binds the interferon α receptor (IFNAR), which initiates the janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway [
44] by phosphorylating JAK [
45]. Phosphorylated JAK recruits STAT, a transcription factor [
44], which is then phosphorylated and dimerizes. Dimerized STAT translocates to the nucleus [
45], which leads to the production of interferon stimulated genes, or ISGs [
44] (
Figure 1). Betaherpesviruses have developed numerous methods of evading this pathway as will be discussed shortly.
Figure 1. Pathogen Sensing, Interferon (IFN), and the JAK-STAT Pathway. Pathogen invasion triggers several pattern recognition receptors (PRRs), including NOD-like receptors (NLRs), RIG-I -like receptors (RIG-I), cGAS, and TLRs (toll-like receptors). These PRRs activate signal transduction pathways that culminate in the production of transcription factors and interferon regulatory factors, which in turn leads to the production of antimicrobial peptides, proinflammatory cytokines, chemokines, and type I IFN. Type I IFN then binds to the interferon α receptor (IFNAR), which phosphorylates JAK. Phosphorylated JAK recruits STAT, which is then phosphorylated, dimerizes, and translocates to the nucleus. Once in the nucleus, STAT upregulates the transcription of ISGs. The HCMV protein pUL145 inhibits STAT, while HHV-6B has been shown to upregulate STAT. The HCMV protein UL26 inhibits ISGs and ISGylation.
3. Immune Cell Responses
The immune system has numerous effector cells. Key cell types include but are not limited to natural killer cells, T cells, and dendritic cells [
94]. These cells have a variety of immune functions ranging from cytokine secretion to antigen presentation and cytotoxic effect [
95,
96]. Betaherpesviruses must successfully contend with each of them in order to successfully establish and maintain an infection.
4. Autophagy and Apoptosis
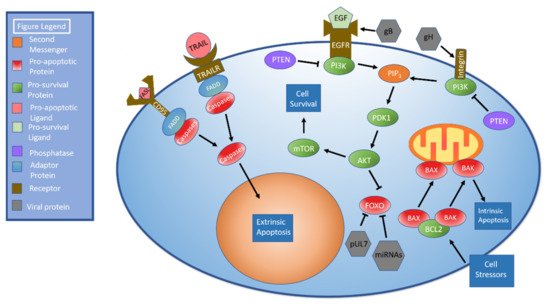
Apoptosis can be triggered by both intra- and extracellular signals. Cell death mediated by external signaling, termed extrinsic apoptosis, is characterized by the binding of several different molecules including FAS/CD95L and TNF related apoptosis inducing ligand (TRAIL). These bind to the receptors CD95 and TRAIL receptors 1 and 2, respectively [
147]. When stimulated, these molecules recruit FADD, which recruits procaspases to create a death-inducing signaling complex (DISC). Once created, DISC facilitates autoproteolytic cleavage and activation of caspases. Activated caspases cleave molecules such as lamin A, poly (ADP-ribose) polymerase (PARP), and inhibitor of caspase-activated DNase (ICAD) to stimulate apoptosis [
148]. The intrinsic pathway is triggered following exposure to stressors such as DNA damage or nutrient deprivation. These stressors cause the proteins BAX and BAK to dissociate from BCL2 and damage the mitochondrial membrane, leading to perforation of the mitochondrial membrane and subsequent release of mitochondrial contents [
149].
Figure 2 details the apoptotic pathways.
Figure 2. AKT and Apoptosis. There are two apoptotic pathways: extrinsic and intrinsic. The extrinsic pathway begins when ligands such as FAS ligand (FASL) or TRAIL bind their corresponding cellular receptors. The adaptor protein FADD is recruited to the receptor. FADD recruits procaspases to create the death-inducing signaling complex (DISC). Once DISC has formed, it facilitates autoproteolytic cleavage and caspase activation. Activated caspases cleave target molecules and stimulate apoptosis. The intrinsic pathway is initiated when the cell is exposed to stressors. The pro-apoptotic proteins BAK and BAX dissociate from BCL2 and disrupt the mitochondrial membrane, resulting in release of mitochondrial contents and apoptosis. The AKT pathway, which can be initiated through either EGFR or integrin stimulation, begins with receptor-mediated activation of PI3K. PI3K activation results in the formation of PIP3, which activates PDK1, which in turn activates AKT. AKT activates mTOR, which promotes cell survival. The human cytomegalovirus (HCMV) protein pUL7 and HCMV miRNAs inhibit FOXO. HCMV gB interacts with the EGFR while gH interacts with integrins to stimulate AKT.
Viruses are obligate intracellular parasites, meaning they require living cells for their survival. Thus, it stands to reason that viruses have strategies to subvert the apoptotic pathway. In fact, Arcangeletti et al., determined that both HCMV and HHV-6A significantly altered the expression levels of numerous apoptosis-associated genes, with HCMV having a greater influence on apoptotic genes than HHV-6A [
150].
Monocytes are essential to HCMV persistence and dissemination [
151]. However, monocytes have a relatively short lifespan of only a few days [
152]. HCMV has been shown to induce monocyte differentiation into longer-lived macrophages that support HCMV replication [
151]. Recent research has shed light on the effect of modulating the AKT signaling pathway on monocyte survival [
153]. In brief, PI3K activation leads to the formation of PIP
3, which activates 3-phosphoinositide-dependent kinase 1 (PDK1). PDK1 phosphorylates and activates AKT. AKT stimulates mTOR signaling, which leads to activation of anabolic pathways as shown in
Figure 2 [
154]. Of note, both EGFR [
155] and integrins [
156] have been shown to stimulate AKT signaling.
Cojohari et al., first described how HCMV induces AKT within 15 min of infection to allow monocyte survival beyond 48 h, which is accomplished by altering expression of the AKT modulating factors PI3K, PTEN, and SHIP1. Moreover, HCMV activation of the AKT pathway was stronger than that induced by macrophage colony stimulating factor (M-CSF) under physiologic conditions [
157]. It was later shown by Peppenelli et al., that the viral glycoproteins gB and gH interacted with epidermal growth factor receptor (EGFR) and αvβ3 integrin, respectively, to stimulate the AKT pathway. These interactions increased myeloid leukemia sequence 1 (Mcl-1) and heat shock protein 27 (HSP27) to deter apoptosis within monocytes [
158]. Mahmud et al. further expanded on this research, determining that gB may have a more significant effect on AKT signaling given that monocytes treated with gB inhibitors alone had lower survivability than those treated with gH alone. While soluble gB and gH could stimulate monocyte survival, monocytes did not survive as well as when infected by HCMV. This may indicate that co-stimulation by gB and gH is necessary to fully potentiate AKT stimulation. Additionally, they showed that gB binds EGFR and that gH binds integrin β1 but not β3 in monocytes. This finding contrasts with the above findings of Peppenelli et al., who found that gH binds αvβ3 integrin. One potential explanation for this discrepancy is that Peppenelli studied fibroblasts while Mahmoud studied monocytes. Interestingly, treatment with either an integrin or EGFR inhibitor blocked signaling through both integrin β1 and EGFR, implying some degree of signal crosstalk. Elimination of this crosstalk blocked HCMV-mediated AKT signaling. EGFR inhibition resulted in abrogated upregulation of MCL-1 and HSP27, which are anti-apoptotic proteins generated by the AKT signaling cascade. The simultaneous activation of EGFR and integrin β1 results in noncanonical activation of AKT signaling. Lastly, inhibition of SHIP was shown to downregulate the pro-survival proteins Mcl-1 and HSP27 [
159]. This contrasts with its normal inhibitory function [
154]; however, aberrant SHIP function has been previously demonstrated in cancer cells [
160].
FOXO is a pro-apoptotic protein family targeted by AKT to promote cell survival [
161] and represents another point at which HCMV can regulate apoptosis. HCMV protein pUL7 stimulates the phosphorylation, translocation to the cytoplasm, and inactivation of FOXO3a through the MAPK pathway. Telomerized human fibroblast cells treated with pUL7 had significantly lower levels of FOXO3a downstream targets BCL2L11 (BIM) and CDKN1B (p27). mRNA expression and protein levels of FOXO3a were unchanged, indicating that regulation occurred via phosphorylation rather than inhibition or degradation, but pUL7 did decrease mRNA levels of the pro-apoptotic protein BCL2L11 [
162].
miRNAs represent another means of FOXO3a targeting and subsequent anti-apoptotic regulation. Transfection of HEK293T cells with HCMV miR-US5-1 and miR-UL112-3p was found to target the 3′ UTR of FOXO3a, which caused FOXO3a transcript and protein levels to decrease [
162].
Autophagy is essential to cellular function. Not only does this process dispose of worn cellular components, but their breakdown is an important source of energy and materials to synthesize new components [
163]. Autophagy is also integral to the normal immune response with functions including but not limited to antigen presentation and pathogen destruction [
164].
Viruses are known to manipulate the autophagic process for their survival and benefit [
164]. Romeo et al., noted autophagosomes, which are involved in the autophagic process, in both infected and nearby uninfected cells as well as an increase in lipidated LC3II, a marker of autophagic flux, in HSB-2 cells after infection with HHV-6A. By comparison, HHV-6B was found to block autophagy in infected Molt-3 cells. Autophagy blockage was confirmed by the relative paucity of autophagolysosomes in both infected and nearby uninfected cells as well as the accumulation of p62, a protein that is normally degraded by autophagy. The two were found to differentially regulate the unfolded protein response [
165], which is associated with endoplasmic reticulum stress. Endoplasmic reticulum stress triggers autophagy, which can help restore the endoplasmic reticulum to its baseline state. If the stressor is too severe, apoptosis ensues [
166]. While HHV-6A increased expression of immunoglobulin heavy chain binding protein (BiP), an anti-apoptotic protein, HHV-6B upregulated C/EBP homologous protein (CHOP), a pro-apoptotic protein. This was accomplished by modulating expression levels of the upstream factors inositol requiring enzyme 1 alpha (IRE1α), activating transcription factor 4 (ATF4), and activating transcription factor 6 (ATF6) [
165].
Autophagy dysregulation has also been shown to play a role in inhibiting dendritic cell formation and monocyte survival in HHV-6B infection. HHV-6B extracted from patients with exanthem subitum was able to inhibit monocyte differentiation into dendritic cells and decrease overall monocyte survival. The addition of sodium 4-phenylbutyrate, which reduces endoplasmic reticulum stress by preventing protein aggregation and acting as chaperone to stabilize folded proteins, attenuated the HHV-6B-mediated inhibition of monocyte differentiation and improved monocyte survival [
167]. A general summary of betaherpesvirus immunoevasive strategies can be found in the table below (
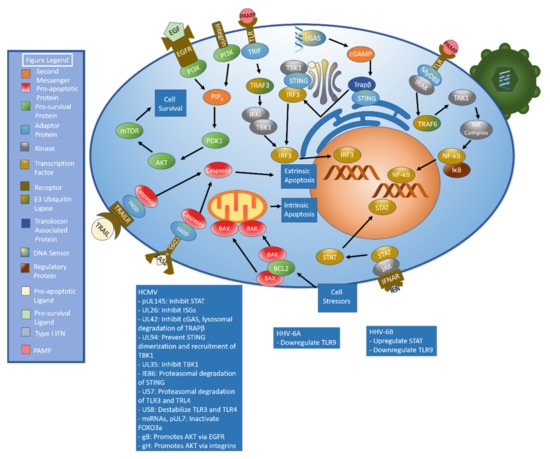
Table 1). A graphical summary of pathways discussed above with a listing of key immunoevasive strategies can also be found below (
Figure 3).
Figure 3. Summary of Key Pathways and Associated Immunoevasive Methods. The immune response to betaherpesviruses begins with the detection of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs), including cGAS and toll-like receptors (TLRs). The cGAS/cGAMP/STING signaling axis results in the production of interferon (IFN) while TLRs can generate both IFN and other defensive compounds. IFN results in STAT activation, which stimulates production of interferon-stimulated genes (ISGs). Betaherpesvirus manipulation of cell survival is an integral element in their success as pathogens. Apoptosis may be stimulated by either intrinsic (BAK/BAX mediated mitochondrial perforation) or extrinsic (FAS ligand/CD95 binding, TRAIL/TRAILR binding) signals. The PI3K/AKT/mTOR signaling pathway represents a major pro-survival cascade. Immunoevasive methods are listed below.
Table 1. Summary of Betaherpesvirus Immunoevasive strategies.
|
Virus
|
Immunoevasive Strategy
|
Associated References
|
|
HCMV
|
UL35: Decrease TBK/IRF3 phosphorylation
|
[61]
|
| |
UL42: Inhibit cGAS’ ability to bind HCMV DNA
Inhibit cGAS oligomerization
Facilitate TRAPβ degradation (lysosome)
|
[56]
|
| |
UL94: Inhibit STING dimerization
Prevent STING from recruiting TBK1
|
[57]
|
| |
IE86: Facilitate STING degradation via proteasome
|
[58,59]
|
| |
US7: Facilitate TLR3 and TLR4 proteasomal degradation
|
[69]
|
| |
US8: Inhibit TLR3 and TLR4 by destabilization
|
[69]
|
| |
IE1: Suppress CpG motifs to avoid ZAP detection
|
[81]
|
| |
UL26: Inhibit ISG15/ISGylation
|
[85]
|
| |
pUL145: Downregulate STAT
|
[90]
|
| |
UL147a: Target MICA for lysosomal degradation
|
[102]
|
| |
UL148a: Target MICA for lysosomal degradation
|
[101]
|
| |
gB: Interact with epidermal growth factor receptor to stimulate the AKT pathway
|
[157,158,159]
|
| |
gH: Interact with αvβ3 integrin to stimulate the AKT pathway
|
[157,158,159]
|
| |
pUL7: Stimulate phosphorylation, translocation to cytoplasm, and inactivation of FOXO3a
|
[162]
|
| |
miR-US5-1 and miR-UL112-3p: FOXO3a inhibition
|
[162]
|
| |
Downregulate CIITA
|
[117]
|
| |
Downregulate MHC class I expression
|
[122]
|
| |
Alter expression of apoptotic genes
|
[150]
|
|
HHV-6A
|
Downregulate IFI16
|
[62]
|
| |
Induce phosphorylation of STAT6
|
[62]
|
| |
Inhibit TLR9 mRNA and protein expression
|
[62]
|
| |
Infect NK cells and alter miRNA and transcription factor expression
|
[24]
|
| |
Modulate HLA class I and class II expression
|
[130]
|
| |
Alter T lymphocyte miRNA upon infection
|
[131]
|
| |
Favor TH2 cytokine profile upon dendritic cell infection
|
[145]
|
| |
Alter expression of apoptotic genes
|
[150]
|
| |
Increase autophagic flux
|
[165]
|
| |
Increase BiP
|
[165]
|
|
HHV-6B
|
Downregulate IFI16
|
[62]
|
| |
Inhibit TLR9 protein levels
|
[62]
|
| |
Inhibit TLR signaling
|
[70]
|
| |
Inhibit autophagy
|
[165,167]
|
| |
Upregulate CHOP via modulating expression of IRE1α, ATF4, and ATF6
|
[165]
|
| |
Inhibit monocyte survival and differentiation into dendritic cells
|
[167]
|
| |
Increase ROS to upregulate PD-L1, influence STAT phosphorylation
|
[93]
|
| |
Downregulate NKG2D ligands
|
[106]
|
| |
Infect NK cells and alter miRNA and transcription factor expression
|
[24]
|
|
HHV-7
|
U21: Targets MCH class I complex for lysosomal degradation, target UBPL1 for lysosomal degradiation, decrease MICA and MICB concentration, post-transcriptional modification of MICA and MICB
|
[109,110,111,133]
|
| |
Slight decrease in cGAS expression
|
[62]
|
5. Latency and Reactivation
Upon infection, betaherpesviruses establish lifelong latency in the human host. The virus can subsequently be reactivated under certain conditions and enter a lytic phase. Latency and reactivation play an important role in betaherpesviruses’ evasion of the human immune system, and there are many facets to the control of these processes [
4].
6. Vaccination Efforts
Betaherpesviruses express numerous glycoproteins that serve as essential components of their fusion machinery [
204,
205,
206]. Recently, these glycoproteins have been assessed as potential targets in vaccination efforts.
Of the betaherpesviruses, efforts to create a vaccine against human cytomegalovirus have been the most robust. As was mentioned previously, HCMV is associated with congenital infection and a relatively high risk of associated birth defects, including but not limited to cognitive impairment, cerebral palsy, bilateral hearing loss, and death [
21,
207]. HCMV infection also causes significant complications in transplant patients, such as transplant-associated vasculopathy, hepatitis, retinitis, infection, and graft rejection [
208,
209].
The HCMV glycoprotein B (gB) has been examined as a potential target for vaccination efforts. Nelson et al., recently examined a lipid nanoparticle vaccine (LNPV) containing a nucleoside modified mRNA transcript encoding full length gB protein in comparison to vaccines containing gB ectodomain (gB protein with antigenic domain 3 removed) with a squalene adjuvant and full length gB with an MF59-like squalene-based adjuvant. Three doses of each vaccine were delivered to New Zealand White rabbits four weeks apart. While the three vaccines were equivalent in their maximal immunogenicity and elicited IgG affinity for gB, the LNPV demonstrated enhanced response longevity relative to the other two. In addition, the LNPV was able to generate antibodies with a broader spectrum of gB peptide binding abilities [
210].
A second effort at targeting gB for vaccine development found that HCMV-neutralizing antibodies primarily targeted antigenic domain 5 [
211]. Using this information, a nanoparticle vaccine was created that increased titers of neutralizing antibodies by 100-fold in comparison to gB extracellular domain vaccine [
211], presumably by increasing the number of copies of antigen presented and by presenting the antigens in a well-ordered manner that may more closely resemble the pathogen surface [
211,
212].
While gB is frequently targeted for HCMV vaccine generation, other promising targets have emerged. The gH/gL/UL128/UL130/UL131 pentamer, a glycoprotein complex required for entry into epithelial and endothelial cells but not fibroblasts [
205], is one such target. A recent series of experiments by Chiuppesi et al., used a modified vaccinia virus ankara (MVA) vector to deliver an antigen including the pentamer complex, the strong T cell stimulator [
213] phosphoprotein 65, and gB. The antigen stimulated a potent immune response involving both humoral and cell-mediated immunity [
214]. A separate study using a guinea pig/guinea pig cytomegalovirus model demonstrated that a disabled infectious single-cycle viral vaccine expressing the pentamer complex was 97% effective in preventing congenital guinea pig cytomegalovirus infection [
215]. Notably, the guinea pig is an accepted animal model for human congenital cytomegalovirus infection [
216,
217]. A more comprehensive review of cytomegalovirus vaccination principles and recent advances in HCMV vaccine technology can be found elsewhere [
218,
219,
220].
While it has not received the same degree of attention as HCMV, there have been some recent efforts to develop a vaccine for HHV-6B. HHV-6 expresses several envelope glycoproteins, including glycoproteins H, L, Q1, and Q2, or gH, gL, gQ1, and gQ2, respectively [
221,
222]. These glycoproteins complex to form a gH/gL/gQ1/gQ2 tetramer that facilitates viral entry by binding CD46 during HHV-6A infection and CD134 during HHV-6B infection [
223,
224]. A recent study by Wang et al., assessed the efficacy of the gH/gL/gQ1/gQ2 tetramer in conjunction with the common vaccine adjuvants aluminum hydroxide gel adjuvant (alum) and D35, a CpG oligodeoxynucleotide adjuvant. N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate was mixed with the D35. A series of three injections given to hSIRPα-DKO mice stimulated the development of both cell-mediated and humoral immunity as measured by cytokine production (specifically IFN-γ and IL-13) and antibody production, respectively [
225]. A PubMed search did not reveal any research into developing a vaccine against HHV-7.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22147503