 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jiahong Lu | + 2748 word(s) | 2748 | 2021-10-11 04:44:07 | | | |
| 2 | Lindsay Dong | Meta information modification | 2748 | 2021-10-27 05:12:35 | | |
Video Upload Options
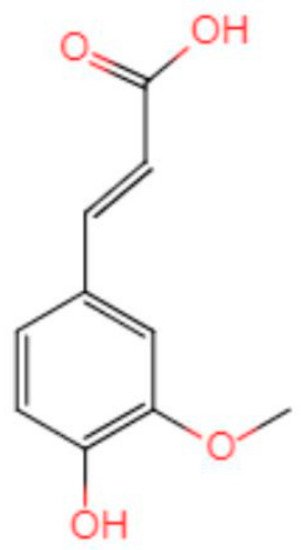
Alzheimer’s disease (AD) is a neurodegenerative disease with a high incidence in the elderly. Many preclinical studies show that a natural product, ferulic acid (FA), displays neuroprotective effects in AD models.
1. Introduction
2. Ferulic Acid in Animal Models of Alzheimer’s Disease
Animal experiments are indispensable for human health science research. In recent years, with the rapid development of biotechnology, the requirements for animal welfare and animal ethics have gradually strengthened. The 3R principle (replacement, reduction, refinement) is one of the representative products. Using systematic reviews to conduct targeted statistical analysis of published animal-related experiments can help researchers make full use of existing data, avoid unnecessary repetitive experiments, optimize existing experimental designs, and reduce cost.
2.1. Mechanism of FA in Anti-AD
The pathogenesis of AD is complex, and the etiology has not been fully elucidated. The current hypothesis involves many aspects such as Aβ toxicity, tau hyperphosphorylation, neuroinflammation, oxidative stress, and abnormal immune function.
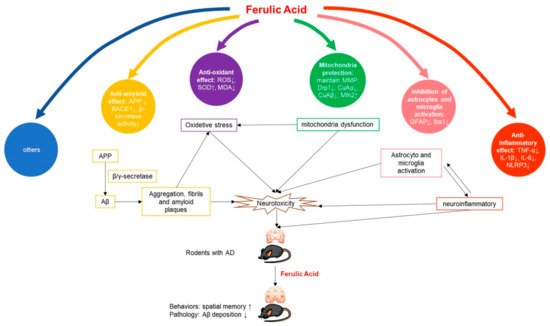
2.1.1. Anti-Amyloid Effect
2.1.2. Anti-Inflammatory Effect
2.1.3. Antioxidant Effect
2.1.4. Mitochondria Protection
2.1.5. Inhibition of Astrocytes and Microglia Activation
2.1.6. Others
2.2. Anti-AD Potential of FA
2.3. Development Perspective
2.3.1. Mechanism Exploration
2.3.2. Structural Modification
2.3.3. Extrapolation to Humans
References
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944.
- Prince, M.J.; Wimo, A.; Guerchet, M.M.; Ali, G.C.; Wu, Y.T.; Prina, M. World Alzheimer Report 2015. The Global Impact of Dementia. An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015; pp. 10–11.
- Shah, H.; Albanese, E.; Duggan, C.; Rudan, I.; Langa, K.M.; Carrillo, M.C.; Chan, K.Y.; Joanette, Y.; Prince, M.; Rossor, M. Research priorities to reduce the global burden of dementia by 2025. Lancet Neurol. 2016, 15, 1285–1294.
- Mathew, S.; Abraham, T.E. Ferulic Acid: An Antioxidant Found Naturally in Plant Cell Walls and Feruloyl Esterases Involved in its Release and Their Applications. Crit. Rev. Biotechnol. 2004, 24, 59–83.
- Smith, B.G.; Harris, P.J. Ferulic acid is esterified to glucuronoarabinoxylans in pineapple cell walls. Phytochemistry 2001, 56, 513–519.
- Yun, L.; Zhao, H.P.; Jing, Z.; Jie, W.; Liao, Z.G. Effect of ferulic acid on learning and memory impairments of vascular dementia rats and its mechanism of action. Acta Pharm. Sin. 2012, 47, 256–260.
- Wu, J.L.; Shen, M.M.; Yang, S.X.; Wang, X.; Ma, Z.C. Inhibitory effect of ferulic acid on neuroinflammation in LPS-activated microglia. Chin. Pharmacol. Bull. 2015, 31, 97–102.
- Kumar, N.; Pruthi, V. Potential applications of ferulic acid from natural sources. Biotechnol. Rep. 2014, 4, 86–93.
- Yan, J.-J.; Jung, J.-S.; Kim, T.-K.; Hasan, A.; Hong, C.-W.; Nam, J.-S.; Song, D.-K. Protective effects of ferulic acid in amyloid precursor protein plus presenilin-1 transgenic mouse model of Alzheimer disease. Biol. Pharm. Bull. 2013, 36, 140–143.
- Mori, T.; Koyama, N.; Guillot-Sestier, M.-V.; Tan, J.; Town, T. Ferulic acid is a nutraceutical beta-secretase modulator that improves behavioral impairment and alzheimer-like pathology in transgenic mice. PLoS ONE 2013, 8, e55774.
- Mori, T.; Koyama, N.; Tan, J.; Segawa, T.; Maeda, M.; Town, T. Combination therapy with octyl gallate and ferulic acid improves cognition and neurodegeneration in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 2017, 292, 11310–11325.
- Mori, T.; Koyama, N.; Tan, J.; Segawa, T.; Maeda, M.; Town, T. Combined treatment with the phenolics (-)-epigallocatechin-3-gallate and ferulic acid improves cognition and reduces Alzheimer-like pathology in mice. J. Biol. Chem. 2019, 294, 2714–2731.
- Qian, W.; Wei-wei, Q.; Jie-wen, Z. Effect of Ferulic Acid on Learning and Memory Impairment by the Repairing of Mitochondrial Fission-Fusion Imbalance in AD Mice. Chin. Pharm. J. 2019, 54, 703–710.
- Huang, H. The Anti-Alzheimer Disease Mechanism of Ferulic Acid as the Main Target of Phosphodiesterase. Ph.D. Thesis, Beijing University of Technology, Beijing, China, 2016; pp. 1–131.
- Mamiya, T.; Kise, M.; Morikawa, K. Ferulic acid attenuated cognitive deficits and increase in carbonyl proteins induced by buthionine-sulfoximine in mice. Neurosci. Lett. 2008, 430, 115–118.
- Tsai, F.-S.; Wu, L.-Y.; Yang, S.-E.; Cheng, H.-Y.; Tsai, C.-C.; Wu, C.-R.; Lin, L.-W. Ferulic acid reverses the cognitive dysfunction caused by amyloid beta peptide 1-40 through anti-oxidant activity and cholinergic activation in rats. Am. J. Chin. Med. 2015, 43, 319–335.
- Yue, W.; Xu, W.; Song, Y.U.; Chun, W. Effects of Ferulic Acid on Oxidative Stress and Apoptosis Related Proteins in Alzheimer’s Disease Transgenic Mice. Nat. Prod. Res. Dev. 2017, 29, 762–766.
- Zafeer, M.F.; Firdaus, F.; Anis, E.; Mobarak Hossain, M. Prolong treatment with Trans-ferulic acid mitigates bioenergetics loss and restores mitochondrial dynamics in streptozotocin-induced sporadic dementia of Alzheimer’s type. Neurotoxicology 2019, 73, 246–257.
- Kim, H.S.; Cho, J.Y.; Kim, D.H.; Yan, J.J.; Lee, H.K.; Suh, H.W.; Song, D.K. Inhibitory effects of long-term administration of ferulic acid on microglial activation induced by intracerebroventricular injection of β-amyloid peptide (1–42) in mice. Biol. Pharm. Bull. 2004, 27, 120–121.
- Beibei, J.; Qin, C.; Qinglin, C. Effect of Ferulic Acid on Learing-memory and Expression of GFAP in the Hippocampus Tissue of Alzheimer’s Disease-like Modle Mice. Acta Laser Biol. Sin. 2011, 20, 484–489.
- Rui, M.; Yi-qing, C.; Qin, C. Effects of ferulic acid on glial activation and inflammatory cytokines expression in the cerebral cortex of Alzheimer’s disease like model mice. Chin. Hosp. Pharm. J. 2018, 38, 50–53.
- Yan, J.J.; Cho, J.Y.; Kim, H.S.; Kim, K.L.; Jung, J.S.; Huh, S.O.; Suh, H.W.; Kim, Y.H.; Song, D.K. Protection against β-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br. J. Pharmacol. 2001, 133, 89–96.
- Cho, J.-Y.; Kim, H.-S.; Kim, D.-H.; Yan, J.-J.; Suh, H.-W.; Song, D.-K. Inhibitory effects of long-term administration of ferulic acid on astrocyte activation induced by intracerebroventricular injection of beta-amyloid peptide (1-42) in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 901–907.
- Ono, K.; Hirohata, M.; Yamada, M. Ferulic acid destabilizes preformed beta-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 2005, 336, 444–449.
- Jagota, S.; Rajadas, J. Effect of phenolic compounds against Abeta aggregation and Abeta-induced toxicity in transgenic C. elegans. Neurochem. Res. 2012, 37, 40–48.
- Yan, R.Q.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.Y.; Gurney, M.E. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999, 402, 533–537.
- Goldsbury, C.; Whiteman, I.T.; Jeong, E.V.; Lim, Y.A. Oxidative stress increases levels of endogenous amyloid-β peptides secreted from primary chick brain neurons. Aging Cell 2008, 7, 771–775.
- Pappolla, M.A.; Chyan, Y.J.; Omar, R.A.; Hsiao, K.; Bozner, P. Evidence of oxidative stress and in vivo neurotoxicity of beta-amyloid in a transgenic mouse model of Alzheimer’s disease: A chronic oxidative paradigm for testing antioxidant therapies in vivo. Am. J. Pathol. 1998, 152, 871–877.
- William, R. Markesbery Oxidative Stress Hypothesis in Alzheimer’s Disease. Free Radic. Biol. Med. 1997, 23, 134–147.
- Butterfield, D.A.; Boyd-Kimball, D. Amyloid β-Peptide(1-42) Contributes to the Oxidative Stress and Neurodegeneration Found in Alzheimer Disease Brain. Brain Pathol. 2006, 14, 426–432.
- Kanski, J.; Aksenova, M.; Stoyanova, A.; Butterfield, D.A. Ferulic acid antioxidant protection against hydroxyl and peroxyl radical oxidation in synaptosomal and neuronal cell culture systems in vitro: Structure-activity studies. J. Nutr. Biochem. 2002, 13, 273–281.
- Bumrungpert, A.; Lilitchan, S.; Tuntipopipat, S.; Tirawanchai, N.; Komindr, S. Ferulic Acid Supplementation Improves Lipid Profiles, Oxidative Stress, and Inflammatory Status in Hyperlipidemic Subjects: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Nutrients 2018, 10, 713.
- Yu, H.; Lin, X.; Wang, D.; Zhang, Z.; Guo, Y.; Ren, X.; Xu, B.; Yuan, J.; Liu, J.; Spencer, P.S.; et al. Mitochondrial Molecular Abnormalities Revealed by Proteomic Analysis of Hippocampal Organelles of Mice Triple Transgenic for Alzheimer Disease. Front. Mol. Neurosci. 2018, 11, 74.
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimers Dis. 2016, 57, 1087–1103.
- Xu, L.L.; Shen, Y.; Wang, X.; Wei, L.F.; Wang, P.; Yang, H.; Wang, C.F.; Xie, Z.H.; Bi, J.Z. Mitochondrial dynamics changes with age in an APPsw/PS1dE9 mouse model of Alzheimer’s disease. Neuroreport 2017, 28, 222–228.
- Reddy, P.H.; Reddy, T.P.; Manczak, M.; Calkins, M.J.; Shirendeb, U.; Mao, P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 2011, 67, 103–118.
- Nitta, A.; Fukuta, T.; Hasegawa, T.; Nabeshima, T. Continuous Infusion of BETA-Amyloid Protein into the Rat Cerebral Ventricle Induces Learning Impairment and Neuronal and Morphological Degeneration. Jpn. J. Pharmacol. 1997, 73, 51–57.
- Westerman, M.A.; Cooperblacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.H.; Carlson, G.A.; Younkin, S.G.; Ashe, K.H. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2002, 22, 1858–1867.
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84.
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Selkoe, D.J. Amyloid β-Protein Dimers Isolated Directly from Alzheimer Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842.
- Gao, C.Y.; Cui, X.Y. Behavioral Study on Learning and Memory Ability of BALB/c and ICR Mice. Acta Lab. Anim. Sci. Sin. 2007, 5, 372–375.
- Liu, G.; Zeng-Yao, H.U.; Yang, S.; Zhou, W.X.; Zhang, Y.X. Comparison of Alzheimer’s disease animal model in BALB/c and Kunming mice by intracerebroventricular injection of β-amyloid. Bull. Acad. Mil. Med. Sci. 2009, 33, 554–557.
- Zhao, Z.; Egashira, Y.; Sanada, H. Ferulic Acid Is Quickly Absorbed from Rat Stomach as the Free Form and Then Conjugated Mainly in Liver. J. Nutr. 2004, 134, 3083–3088.
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 13, 1763.

