Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Igor Bychkov | + 2075 word(s) | 2075 | 2021-10-14 08:55:40 | | | |
| 2 | Amina Yu | -6 word(s) | 2069 | 2021-10-28 04:03:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bychkov, I. Complex Transposon Insertion, Novel Cause of Pompe Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/15426 (accessed on 27 July 2026).
Bychkov I. Complex Transposon Insertion, Novel Cause of Pompe Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/15426. Accessed July 27, 2026.
Bychkov, Igor. "Complex Transposon Insertion, Novel Cause of Pompe Disease" Encyclopedia, https://encyclopedia.pub/entry/15426 (accessed July 27, 2026).
Bychkov, I. (2021, October 26). Complex Transposon Insertion, Novel Cause of Pompe Disease. In Encyclopedia. https://encyclopedia.pub/entry/15426
Bychkov, Igor. "Complex Transposon Insertion, Novel Cause of Pompe Disease." Encyclopedia. Web. 26 October, 2021.
Copy Citation
Pompe disease (OMIM#232300) is an autosomal recessive lysosomal storage disorder caused by mutations in the GAA gene. According to public mutation databases, more than 679 pathogenic variants have been described in GAA, none of which are associated with mobile genetic elements. In this article, we report a novel molecular genetic cause of Pompe disease, which could be hardly detected using routine molecular genetic analysis. Whole genome sequencing followed by comprehensive functional analysis allowed us to discover and characterize a complex mobile genetic element insertion deep in the intron 15 of the GAA gene in a patient with infantile onset Pompe disease.
SVA
L1
transposable elements
transcription termination
missplicing
retrotransposon
transposon insertion
lysosomal storage disease
functional analysis
1. Patient’s Summary
The patient is a 1.5-month-old male from the fifth birth in a consanguineous marriage. The family also had one miscarriage at 7 weeks, two children with hypertrophic cardiomyopathy, who died before 1 year, and one healthy 9-year-old child (Figure 1A). Medical examination of the patient revealed muffled heart sounds, suspected hypertrophic cardiomyopathy on echocardiography, elevated liver aminotransferases and creatine kinase activity and reduced tendon reflexes in the lower limbs. Based on the anamnesis and the family history, PD was suspected. The subsequent biochemical analysis revealed the decreased activity of alpha-1,4-glucosidase—0.130 μmol/L/h (normal range 1–20), which is the marker of PD.
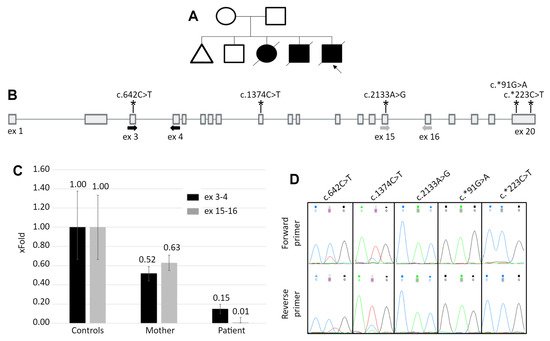
Figure 1. The patient’s pedigree and results of the white blood cells mRNA analysis. (A) The patient’s pedigree. Blacked out symbol—patient with the PD phenotype; crossed out—deceased patient; triangle—miscarriage. (B) The scheme of the GAA gene. Bold arrows indicate location of the primers for qPCR and asterisks indicate the location of heterozygous variants, identified in the mother’s DNA. (C) The results of qPCR for the GAA cDNA with primers spanning exons 3–4 and 15–16. Controls—mean and standard deviation for 5 healthy control samples. For the mother’s and the patient’s samples the standard deviation was calculated from technical replicates. (D) Sanger chromatograms of the mother’s cDNA fragments containing heterozygous variants. The allelic imbalance demonstrates the similar to the qPCR data, expression pattern.
2. Molecular Genetic Analysis
To establish the molecular genetic diagnosis of PD, we analyzed the exons with about 200 b.p. of adjacent introns and the promoter region of the patient’s GAA gene by Sanger sequencing but did not reveal any rare suspicious variants.
To identify the possible splicing alterations, which could be caused by the deep intronic variants, the RNA extracted from the patient’s white blood cells was analyzed. After cDNA synthesis, the whole coding sequence of the GAA mRNA was amplified by overlapping fragments, visualized by agarose gel electrophoresis and Sanger sequenced. No additional PCR products were detected compared to the control sample and Sanger sequencing also did not reveal any abnormalities.
The other considered causes of the altered GAA function were the mutations located in regulatory sequences, disruption of topologically associated domains or translocation of the GAA locus in the region of condense heterochromatin, that lead to severe reduction of gene’s expression. To quantitatively analyze the GAA expression, qPCR primers targeting the 5′ region (3–4 exons) and the 3′ region (exons 15–16) of the GAA cDNA were designed. The results of the qPCR revealed that the patient’s GAA expression is reduced to 15% of the control at the 5′ region and to 1% at the 3′ region (Figure 1C).
According to the GIS ChIA-PET track of the UCSC genome browser (https://genome.ucsc.edu/) (revised on 15 July 2021), the GAA promoter has a strong three-dimensional interaction with the EIF4A3 promoter and is probably common for these two genes. Assuming that the reduced activity of the GAA promoter could affect the expression of EIF4A3, we measured its expression by qPCR and revealed that it did not differ from the control. This observation suggested that the causative mutation most probably is located in the GAA gene’s body.
At this step the patient’s cDNA was used up and no more mRNA or fresh blood were available because the patient died. Therefore, the mother’s blood was obtained, and all subsequent mRNA analyses were carried out on the mother’s sample.
As the mother can be the carrier of the allele with reduced expression, we performed the analysis of the allelic imbalance at the GAA’s mRNA level. The sequencing of the GAA exons amplified from the mother’s DNA revealed five heterozygous variants. The sequencing of the cDNA fragments containing these variants demonstrated the characteristic allelic imbalance, similar to the expression pattern in patient’s cDNA—partial loss of zygosity for two variants in the 5′ half of the gene and complete loss of zygosity for three variants in the 3′ half (Figure 1D).
At the next step, whole genome sequencing was performed using the patient’s DNA. Automatic annotation of the genome using the standard variant calling protocol did not reveal any suspicious variants in the GAA gene, but the visual inspection of the alignment file identified the duplication of 15 b.p. (NC_000017.11:g.80,114,172_80,114,186dup) within the intron 15 of GAA. In addition, the discordant reads flanking this duplication were identified, mapping at a chromosome 20 region that contains polymorphic SVA element named CPX-1 (NC_000020.11:2,822,517-2,825,763) [1] (Figure 2A). SVA is a mobile genetic element, which transcribes from DNA locus and uses the RNA molecule intermediate during transposition. Being nonautonomous, SVA requires L1 proteins for integrating into the genome by target-primed reverse transcription with the formation of target site duplication near the integration site “attaaaa”, which is located in the GAA intron 15 [2][3]. The intron 15 of GAA, in turn, does not contain any polymorphic transposable elements according to the database of retrotransposon insertion polymorphisms dbRIP (http://dbrip.brocku.ca/searchRIP.html) (revised on 15 July 2021).
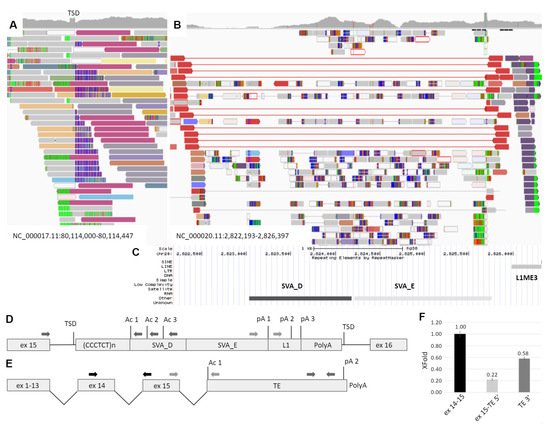
Figure 2. Identification and characterization of the TE insertion. (A) The IGV browser window representing the discordant and split reads mapped to the intron 15 of the GAA gene. TSD—target site duplication formed during TE integration and represented by the increased coverage. (B) Reads mapped to the region of the transposition origin at chr20. The red reads flank the polymorphic CPX-1 transposon presented in patient in heterozygous state. Dark blue reads are discordant, and their pairs are mapped to the GAA intron 15 (dark red reads at Figure 1A). (C) The RepeatMasker track of the UCSC browser aligned with IGV and representing the mobile genetic elements in the origin of transposition. (D) The scheme of TE insertion in GAA intron 15 at the DNA level. Ac 1–3—several strong acceptor splice sites; pA 1–3—several transcription termination signals. (E) The scheme of the chimeric GAA transcript isoform with exonization of TE. (F) The results of transcript-specific qPCR of the mother’s cDNA sample. The relative amounts of different parts of the chimeric GAA transcript isoform are presented.
Thus, the retrotransposition of the SVA element from chromosome 20 was suspected. The Sanger sequencing of the insertion boundaries established the precise coordinates of the probable retrotransposition origin at NC_000020.11:2,823,027-2,826,302. According to the RepeatMasker track of the UCSC browser, this region contains TEs of the SVA_D, SVA_E and L1ME3 classes (Figure 2B). The complete 3394 b.p. insertion was also amplified from the patient’s DNA with the primers located in the introns of GAA and was sequenced by NGS. The obtained sequence matches the reference by 98.95% (Supplementary file).
3. Study of Mechanism of the TE Insertion Molecular Pathogenesis
As the GAA intron 15 already contains TE of various types, the pathogenic effect of the novel TE insertion was not obvious. One of the mechanisms by which the cell suppresses the TE activity in its genome is the methylation of the corresponding locus. To identify whether changes in the methylation of the GAA gene are the cause of reduced expression, we performed bisulfite sequencing of two CpG islands located near the exon 1, exons 4–5, and two regions located in intron 15. All of the analyzed CpG pairs in patient’s DNA around the exon 1 were unmethylated, and CpG pairs in three other regions were methylated, which was also observed in the control DNA and represents the normal methylation pattern of the genes.
The vast majority of pathogenic TE insertions, reported as a cause of human diseases, are located in exons, alter splicing sites or serve as a source of cryptic splice sites, causing exonization [4]. As the identified SVA/L1 element is located in the forward orientation relatively to the gene and no exonization or other events were detected during amplification of patient’s cDNA fragment spanning exons 15–16, we hypothesize, that the TE sequence could be spliced with the GAA mRNA as the last exon and terminate the transcription. Therefore, we performed PCR using the mother’s cDNA with primers located in GAA exon 15 and in the 5′ region of transposon (Figure 2C dark grey arrows). This region consists of Alu element in the reverse orientation with several splicing acceptor sites of medium strength (according to https://www.fruitfly.org/seq_tools/splice.html (accessed on 15 July 2021)). Sanger sequencing of the PCR products revealed that the GAA exon 15 was spliced with TE due to activation of the acceptor site composed of the AG dinucleotide located at the end of CCCTCT repeats, serving as the polypyrimidine tract and the branch point located in the GAA intron. It is worth noting that this AG dinucleotide was formed due to the indel, corresponding to NC_000020.11:2,823,108-2,823,110delGATinsCAA, which is absent in the reference sequence.
To determine the 3′ end of the chimeric mRNA isoform we performed the rapid amplification of the cDNA ends (3′ RACE) using primers located in L1 and SVA_E regions of TE (Figure 2C light grey arrows). Sanger sequencing of the PCR products revealed the 3′ end of the TE sequence with about 30 b.p. polyA tail and polyA site located 29 b.p. downstream of the polyadenilation signal TATAAA located in the L1 element (Figure 2—p(A)2).
To quantitatively estimate the amount of chimeric mRNA and to determine whether the TE sequence exonization is the main cause of the patient’s GAA dysfunction, we applied the real-time PCR with primers designed to amplify three targets: the junction of exons 14–15 of the GAA mRNA, the junction of the GAA exon 15 and the 5′ region of TE sequence and the 3′ end of TE sequence. The corresponding amplicons were cloned into plasmid vector, which was used as a reference sample for ddCq calculation. The relative amount of the exon 15-TE 5′ and TE 3′ molecules normalized to the expression of exons 14–15 was 0.22 and 0.58, respectively. In addition, no significant amplification of exon 15-TE 5′ and TE 3′ target was detected in five control samples.
The results of the allelic imbalance analysis for the c.642C>T and c.1374C>T variants in the mother’s cDNA demonstrated that the TE-containing isoform is reduced to about 30% of the WT isoform (Figure 1D). So, if we consider the allelic imbalance analysis data, the amount of the TE-containing isoform should be about 25% (66.6% (total GAA mRNA level—50% + 1/3*50%) / 16.6%) of total GAA isoforms in the mother’s cDNA. This is in a good agreement with the data from chimeric isoform measurement for ex 15-TE 5′—22%. The most probable reason for high TE 3′ expression (58%) is that the cDNA was synthesized using oligo(dT) primers, which cause the 3′ end bias, especially in the chimeric isoform, containing about 2000 b.p. of the GC-rich sequence between the TE 3′ target and the reference ex 14–15 target. The 3′ end bias is also the reason for using primers for the GAA exons 14–15 instead of exons 2–3 for the chimeric isoform measurement, as they are located as close as possible to the GAA-TE junction.
Together, these results strongly suggest that the exonization of the ~3137 b.p. TE sequence using strong acceptor site right downstream of CCCTCT repeats and polyA signal in the 3′ end of the TE with subsequent termination of transcription is the main deleterious consequence of this insertion.
4. Conclusions
The novel type of mutation, associated with infantile-onset PD. The whole genome sequencing followed by comprehensive functional analysis allowed us to discover and characterize the deleterious effect of the complex mobile genetic element insertion deep in the intron 15 of the GAA gene. This study draws the attention of researchers to the need for detailed analysis of genome sequencing data for the presence of footprints of TE insertions. Currently there is a wide range of freely available bioinformatics algorithms that should be implemented into standard variant calling protocols to detect this underestimated type of mutations. The functional genomics in turn have a wide range of available and effective methods, which could be further used to establish their pathogenicity.
References
- Bennett, E.A.; Coleman, L.E.; Tsui, C.; Pittard, W.S.; Devine, S.E. Natural Genetic Variation Caused by Transposable Elements in Humans. Genetics 2004, 168, 933–951.
- Kojima, K.K. Different integration site structures between L1 protein-mediated retrotransposition in cis and retrotransposition in trans. Mob. DNA 2010, 1, 17.
- Luan, D.D.; Korman, M.H.; Jakubczak, J.L.; Eickbush, T.H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell 1993, 72, 595–605.
- Hancks, D.C.; Kazazian, H.H. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9.
More
Information
Subjects:
Genetics & Heredity
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
778
Revisions:
2 times
(View History)
Update Date:
28 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No