Alzheimer’s disease (AD) is one of the most prominent neurodegenerative diseases, which impairs cognitive function in afflicted individuals. AD results in gradual decay of neuronal function as a consequence of diverse degenerating events. Several neuroimmune players (such as cytokines and growth factors that are key players in maintaining CNS homeostasis) turn aberrant during crosstalk between the innate and adaptive immunities. This aberrance underlies neuroinflammation and drives neuronal cells toward apoptotic decline. Neuroinflammation involves microglial activation and has been shown to exacerbate AD. Cytokines are non-structural proteins within the molecular weight range of 8000–40,000 Da. They can be described as inflammatory peptides aiding the immune defense response. The majority of nucleated cells are capable of synthesizing cytokines but they are predominantly produced by macrophages/microglia and lymphocytes.
1. Introduction
Neurodegeneration has been a puzzle gradually elucidated by the progress of ample research and the investigation of dementia and progressive cognitive decline. Dementia which is marked by the affliction of Alzheimer’s disease (AD), is understood as the decline in memory and other fundamental cognitive functions. AD is the most commonly occurring neurodegenerative disease in the world. AD has been extensively characterized by the gradual decline of neuronal health. Neurotoxins, TAU protein neurofibrillary tangles, amyloid-beta (Aβ) plaque accumulation in mature neuron phenotypes
[1][2][3][4][5], mitochondria dysfunction (fusion-fission imbalance)
[6][7], and neuroinflammation collectively involves in neurodegeneration in AD
[8][9][10][11]. Mitochondrial dysfunction results in the accumulation of harmful reactive oxygen species (ROS), which subsequently trigger CNS apoptotic decline
[7]. Neuroinflammation is mainly governed by the actions of cytokines, chemokines, and growth factors, which play key roles in neurodegeneration
[8][9][10]. These aberrancies have been widely reported as fundamental hallmarks of AD and its pathological quantification
[12][13].
Cytokines are non-structural proteins within the molecular weight range of 8000–40,000 Da. They can be described as inflammatory peptides aiding the immune defense response. The majority of nucleated cells are capable of synthesizing cytokines but they are predominantly produced by macrophages/microglia and lymphocytes
[14]. These cells can in turn also respond to and interact with cytokines. Cytokines can be grouped into certain classes based on their biological activities which could be pro-inflammatory or anti-inflammatory. The biological activities of cytokines are vast and range from cell proliferation to apoptosis and from cell differentiation to inflammatory responses. Cytokines are also termed lymphokines since they are primarily involved in the differentiation of different types of T lymphocytes
viz. T helper cells, and T regulatory cells from undifferentiated cells
[15]. Many of these proteins, for example, interleukins (ILs), interferons (INFs), tumor necrosis factors (TNFs), and certain growth factors are produced by neurons and glial cells of the brain in the event of neuroinflammation. Levels of IL-1α, IL-1β, IL-6, TNF-α, IFN-α, macrophage colony-stimulating factors (MCSFs), IFN-α and IL-8 receptor type B are enhanced in blood and cerebrospinal fluid (CSF) in AD patients. Nerve growth factors (NGF), growth-promoting properties of APP, vascular endothelial growth factor (VEGF) also play vital roles in the pathophysiology of AD. Growth factors are proteins by nature and support the survival of cells within the nervous system. Moreover, they are vital players for the proper development of the brain. In the CNS and PNS, they stimulate axonal growth and regulate the growth of different kinds of cells.
AD is named after German psychiatrist and neurologist Alois Alzheimer
[16]. In 1906, the doctor noted some peculiar findings in the brain of a patient who passed away after suffering from memory loss, disorientation, paranoia, and unpredictable behaviors. AD causes a gradual decline in cognitive processes starting with mild cognitive impairment (MCI) reaching a stage of severe irreversible loss of cognition and functionality (
Table 1). AD, by nature, is an insidious, progressive, and degenerative disorder. Given the fact that the improvements in medical science considerably improve the quality of life and increase life expectancy in afflicted individuals, a longitudinal study that began with a cohort of normal subjects revealed a higher incidence of AD in women compared to men with the largest incidence in age group ≥ 85 (95% CI 5.01 to 8.38)
[17], and epidemiological studies of the prevalence of AD show a positive correlation with increasing age
[18]. AD invariably starts from the hippocampus (responsible for new memory generation) making anterograde amnesia a primary symptom of the disease. As neurofibrillary tangles start to spread outward towards the frontal lobe, dementia is followed by progressive speech problems, mood imbalance, and inability in decisions making
[19]. Several genes including the senilins, SORL1, APP, and ApoE4 were found to play crucial roles in the onset and progression of AD
[19]. Early AD onset is generally familial, while late AD onset is largely related to SORL1. From the viewpoint of pathophysiology, AD is characterized by intracellular neurofibrillary tangles and extracellular senile plaques. Assessment of Instrumental activities of daily living in a geropsychiatry clinic revealed that impairment and memory loss was higher in patients with mild cognitive impairment (MCI) (
n = 66) compared to control subjects (
n = 61)
[20]. During the course of AD progression, individuals begin to experience cognitive decline prior to clinically diagnosed MCI. In a longitudinal study by Cloutier et al., compared to healthy controls who did not progress to an MCI diagnosis, individuals who were previously healthy and later expressed cognitive impairment showed different patterns of impairment years prior to an MCI diagnosis and escalating severity of decline was observed over time
[21]. The incidence of neuropsychological decline constituting memory loss, episodic cognitive decline, and executive function decline 12 years before MCI diagnosis indicate that neuroinflammation is present in neurodegeneration that leads to AD prior to diagnosable MCI
[22]. Brain hypometabolism map PET scan analysis corroborated that the activation of microglial regional clusters in the brains of individuals is predominantly involved in the transition from healthy status to dementia
[23], which divulges the involvement of inflammation in neurodegeneration leading to AD.
Table 1. Stepwise progression of AD.
| Serial. No. |
Stages |
Pathological Symptoms |
| 1 |
Early onset AD/MCI |
Impairment of non-memory features of cognition, difficulty in word finding, decline in reasoning/judgement. |
| 2 |
Mild AD |
Loss of spontaneity, memory loss, anxiety, aggression, restlessness, altered personality, misplacing items. |
| 3 |
Moderate AD |
Confusion, attention deficit, continuous cognition problems, impulsive behavior, delusion, paranoia, hallucination, recognition problem. |
| 4 |
Severe AD |
Severe dementia, continued cognitive decline, seizures, functional limitations, lack of bowel/bladder control, weight loss, skin infection, swallowing difficulty, enhanced sleep time, brain atrophy. |
Identification and elucidation of the roles of cytokines and their co-associating factors, such as growth factors, in the immune system and in response to the pathogenesis of AD, is a key step to explore their potentials for therapeutic interventions. This review aims to analyze research data, prior AD-related research, and affiliations between connected fates of inflammatory and immune responses of AD, to help identify the role of cytokines and key growth factors implicated in AD.
2. Immune Response in AD: Role of Cytokines
Cytokines mediate cell functioning, cell signaling behaviors, and neuro-immune activity and are classified by the actions that they solicit. During AD immune response, such cytokines include pro-inflammatory cytokines, anti-inflammatory cytokines, and cytokines that are known to inhibit virus replication. These cytokines can activate macrophages, B-cells, T-cells, and mast-cells and constitute a cytokine network in the brain. In AD, certain cytokines are involved in the immune responses that precede and stimulate the actions of other cytokines in the innate neuroimmune inflammatory reactions. It was observed in AD consequent of aberrant pathologies in the brain and concomitant to CNS insults that include neurotoxicity, accumulation of Aβ senile plaque, and TAU pathologies (
Table 2). IL-1α containing plasmids were analyzed in IL-1 cDNA clones by the hybrid selection of biologically active mRNA that resulted in abundant IL-1 expression in LPS-stimulated macrophages
[24].
Table 2. Changes mediated by cytokines and growth factors within CNS.
| Serial No. |
Mediators |
Functions |
References |
| 1 |
IL-1α |
Increases α-secretase, decreases amyloidogenic processing, increases sAPPα |
[24][25][26] |
| 2 |
IL-1β |
Increases APP mRNA, increases α-secretase and γ-secretase, downregulates β-secretase, upregulates TAU mRNA |
[27][28][29] |
| 3 |
IL-4 |
Upregulates Aβ production, increases p-TAU |
[29][30] |
| 4 |
IL-6 |
Upregulates APP mRNA, increases p-TAU |
[10][31] |
| 5 |
IL-8/CXCL8 |
Upregulates γ-secretase activity by increasing substrates C83 and C99 |
[32][33] |
| 6 |
IL-10 |
Favors Aβ deposition |
[10][34][35] |
| 7 |
IL-18 |
Increases APP, upregulates both β-secretase and γ-secretase, increases Aβ formation |
[10][36][37] |
| 8 |
TNF-α |
Upregulates APP mRNA, upregulates both β-secretase and γ-secretase, increases sAPPβ |
[10][35][38] |
| 9 |
IFN-γ |
Upregulates APP intracellular domains, upregulates both β-secretase and γ-secretase, increases Aβ deposition |
[39][40][41][42] |
| 10 |
TGF-β1 |
Increases APP mRNA, increases Aβ deposition |
[10][41][42] |
| 11 |
CCL2 |
Increases Aβ formation and deposition |
[43][44] |
| 12 |
CCL3 |
Upregulates β-secretase, increases C99, increases Aβ deposition |
[44][45] |
| 13 |
CCL5 |
Upregulates β-secretase, increase C99, increases Aβ deposition |
[45][46] |
| 14 |
CXCL10 |
Decreases Aβ deposition |
[33][47] |
| 15 |
CX3CL1 |
Decreased Aβ deposition, upregulated p-TAU |
[48][49] |
| 16 |
VEGF |
Upregulates expressions of monocytes and macrophages, increases proliferation of endothelial cells |
[50][51][52] |
| 17 |
FGF |
Attenuates Aβ related pathologies |
[51][53] |
| 18 |
NGF |
Increases degeneration leads to loss of cholinergic nerve endings in cortex and hippocampus |
[54][55] |
| 19 |
BDNF |
Upregulates sAPPα, promotes non-amyloidogenic pathway, astrocyte activation, improved memory performance |
[56][57] |
| 20 |
GDNF |
Neuroprotection |
[54][58] |
| 21 |
GCSF |
Induces neurogenesis |
[59][60] |
| 22 |
Stem cell factor |
Maintains hematopoietic brain support, neurogenesis |
[61][62] |
| 23 |
SDF |
Neurogenesis, inflammatory disruption of BBB |
[63][64] |
| 24 |
CXCR4 |
Ligand for SDF-1 |
[63][65] |
| 25 |
Angiopoeitins |
Angiopoeitin-1 prevents neuronal apoptosis, Angiopoeitin-2 promotes neurogenesis via migration of neural progenitor cells |
[66][67][68] |
| 26 |
Neurotrophin-3 |
Upregulates neuronal apoptosis inhibitory protein 1, limits cleavage of caspases 3, 8 and 9 |
[69][70] |
| 27 |
Neurotrophin-4 |
Regulates TAU dephosphorylation |
[69][71] |
| 28 |
TrKA |
Receptor protein for β-NGF |
[72][73] |
| 29 |
TrKB |
Receptor protein for brain derived neurotrophic factor and neurotrophins |
[72][74] |
| 30 |
TrKC |
Receptor protein for neurotrophin-3 |
[72][75] |
| 31 |
p75 |
Neurotrophin receptor protein, regulates phosphorylation of TAU |
[70][71] |
Of the classes of cytokines that are implicated in AD, specialized groups of cytokines are differentiated by the availability of their receptors expressed on the cell surface of implicated cell types and the condition of the genes that regulate these receptors. Cytokines play a major role in routine neurological activities of the CNS in the transfer and reception of chemical cues that confer instructions on cell actions and reactions. Chemotactic cytokines that function as chemoattractant cytokines, such as IL-8 and IP-10/CXCL10 may experience N-terminal proteolytic alteration after being secreted.
2.1. Immune System in AD and Cytokines
At the beginning of neurodegeneration, the immune reactions trigger macrophage activation (predominantly M2 and sometimes M1)
[76]. These macrophages secrete chemical messengers in interneuronal communications and develop autoimmune neurotoxicity including those reactions that lead to neuroinflammation and escalation of AD. The immune system employs cytokines, which play a major role in immune responses following the activation of microglia in the pathology of AD. Cytokines determine the mechanisms and reactions that take place in the immune system in response to abnormal changes in the neurons. These trigger the recruitment of other defensive cells including neutrophils and macrophage progenitor cells.
In the case of AD, Aβ originating from APP trigger the rest of the pathologies. Aβ outside the neurons and neurofibrillary tangles inside the neurons make up for the development of AD
[77][78]. Aβ further produces immune response activating complement systems. In CNS, the immune system is programmed to functionally respond to pathological changes such as those presented by the progression of AD
[76]. The immune system activation observed in AD is labelled as neuroinflammation
[79]. Herein, misfolded and aggregated proteins i.e., Aβ act through danger-associated molecular pathways (DAMP) to bind pathogen recognition receptors such as CD14, CD36, α6β1, integrin, and toll-like receptors (TLRs)
[80]. These, in turn, control functions of ROS, NO, IL-1β and TNF-α. It has been experimentally shown that, contrary to antiquated conclusions about neuroinflammation, observed in MCI, early, and late AD onset are initiating events predominantly driven by the CNS resident immune cells, such as microglia and perivascular myeloid cells
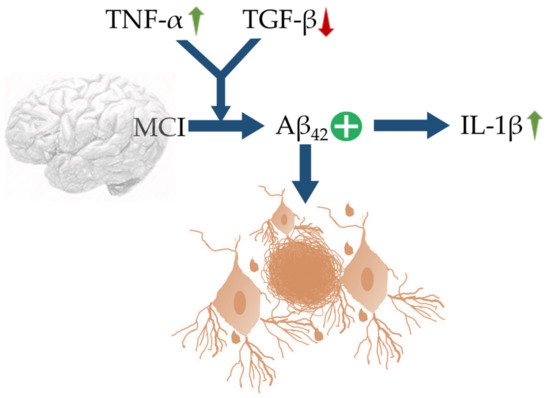
[79]. An up-regulation of TNF-α with concomitant suppression in TGF-β synergize Aβ42 deposition in MCI, which further trigger neuroinflammation via recruiting IL-1β (
Figure 1). Genetic variants and transcription factors also determine the expression of activated microglia in the pathological environment. Damaging or degenerating neurons give off signals acting as a form of microglial control switch that stimulates microglia which could become cytotoxic from the reactive intermediates solicited such as pro-inflammatory cytokines
[81]. In response to a change in homeostasis, microglia must first be activated, changing it from a static to a primed state. Changes in infiltrating monocytes that support CNS immune response in the parenchyma and neuronal progenitor granule crossing the BBB might be a hallmark for early detection of AD and propensity of inflammatory response and neurodegeneration
[82]. Asymmetrical changes in serum and plasma levels of cytokines may indicate changes in early cytokine levels widely reported in macrophage precursor cells that may confer a greater risk of developing neurodegeneration and abnormal macrophage morphology.
Figure 1. Schematic representation of MCI, linked with up-regulation of TNF-α and decrease in TGF-β characterized by upregulation of IL-1β and Aβ42 expressions. The blue arrows (↑) indicate downstream cellular events, upward green arrows (↑) indicate upregulation, downward red arrow (↓) indicates down-regulation, and plus sign (+) indicates enhanced activity.
2.2. Roles of Cytokines in Autophagy
Aβ burden has been revealed to be positively correlated with age
[50] and exacerbated by oxidative stress, such as GAPs that promote the generation of ROS
[53] that perturb brain health
[83][84][85]. Glycation end products that confer oxidative stress in AD, which was found to be heavily associated with ApoE in its dimeric form greater than its monomeric form at Aβ accumulation site
[54]. An increase of ApoE can lower the Aβ
40–42 turnover rate on greater cognitive decline in AD
[56]. The same has also been found to negatively influence or disturb autophagy by disrupting autophagosome formation
[58]. This, in turn, leads to greater deterioration of neuronal health in AD pathology. Autophagy is critical for Aβ clearance and important in the maintenance of homeostasis in the CNS. In concert with dysfunction of autophagy, mitophagy was observed to express excessive fragmentation, decline in synaptic integrity
[59], and an imbalance of mitochondrial dynamics
[60][61]. Dysfunction of autophagy/mitophagy indicates a notable neuroinflammatory pathology and involvement of cytokines. IL-1β and IFN-γ (which are known to be expressed in AD pathogenesis) exposure to primary rat β-islet cells hindered autophagy resulting in cell apoptosis
[63] and additionally, IL-1β was reported to modulate microglia autophagy in LPS cultures in the presence and absence of Aβ42
[66][86]. This evidence suggests that IL-1β and IFN-γ maintain control of inflammation in AD via lysosomal pathway and initiation of phagophore assembly.
2.3. Cytokines and BBB
There exists a definite correlation between brain cytokine levels and neuropsychiatric disorders. Right at this point, selectivity, and integrity of BBB to cytokines become important. Cytokines are pleiotropic, hence their release, unlike hormones has more complicated effects on the regulation of neurotransmission. Cytokines can cross BBB, activate free calcium, and by disrupting the compartmental model of brain calcium homeostasis, compromise the integrity of BBB
[87]. Many cytokines can pass through BBB directly
[88]. Interestingly, glial cell-derived neurotrophic factors bypass the BBB by simple diffusion through circumventricular organs. Whereas passage of IL-1α, IL-6, and TNF-α involves saturable influx transport through retrograde axonal transport system
[87][89]. TNF-α, a downstream cytokine of chemokine IP10, decreases tight junction proteins leading to the destruction of endothelial tight junctions of BBB to affect its permeability
[90]. On the other side, inhibition of mTOR hyperactivity has been reported to protect the integrity of BBB in AD
[91]. Therefore, BBB dysfunction brings about early aging in the brain paving the way for AD and other neurodegenerative disorders.
3. Role of Cytokines and Chemokines in Neuropsychiatry
The study of cytokines to understand the pathophysiology of neuropsychiatric disorders such as dementia, anxiety, and delirium has been pioneered by Dr. M. Maes who first linked vegetative symptoms with enhanced presence of IL-1, IL-6, and haptoglobin
[87][92]. Chemokines regulate the migration of microglia and the recruitment of astrocytes to the sites of inflammation. Cytokines may act in an autocrine, paracrine, or endocrine fashion and generally are upregulated at sites of Aβ plaques. Aβ peptides mediate cell mediators, such as monocytes are also responsible for the generation of IL-8, monocyte chemoattractant protein 1 (MCP1), MIP1α, and MIP1β. LPS stimulates astrocytes to secrete cytokines including IL-6 and TNF-α, activates astrocytoma cells to secrete IL-6 and IL-8 and monocytes to secrete IL-8 under the influence of Aβ peptides
[93]. Synergistic activity of cytokines has also been reported along with Aβ peptides e.g., TNF-γ synergizes with Aβ to enhance secretion of TNF-α and reactive nitrogen species
[38]. IL-1β displays pro-inflammatory actions via MEK 1/2, JNK-activated α-secretase cleavage and upregulated a disintegrin and metalloprotease (ADAM)-17/TNF-α converting enzyme (TACE) pathway to increase sAPPα secretion
[94]. On the contrary, IL-1β can also serve as an anti-amyloidogenic factor by decreasing sAPPβ and amyloidogenic Aβ fragment levels by reducing β-secretase cleavage
[95]. It was also suggested that increased Aβ clearance by microglia in models of sustained IL-1β neuroinflammation could involve Th2 cytokines, such as IL-4
[29]. Moreover, a feedback signalling loop between Aβ and IL-1β was also proposed in which Aβ can induce the production of IL-1β
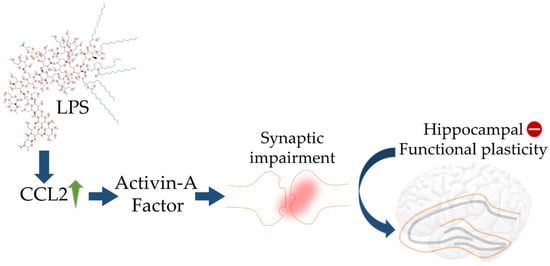
[96]. The migration of astrocytes to Aβ plaques is promoted by chemokines CCL2 and CCL3, which are generally released by activated microglial cells. Upregulation of CCL2 by LPS was found to promote synaptic impairment through recruiting activin A leading to loss of hippocampal plasticity (
Figure 2).
Figure 2. Schematic diagram showing impact of LPS on elicited CCL2 activity in turn leading to aberrant hippocampal plasticity. The blue arrows (↑) indicate downstream cellular events, upward green arrow (↑) indicates upregulation, and minus sign (−) indicates decreased activity.
Important pathways involved in the pathogenesis of AD include the amyloid cascade hypothesis, TAU hypothesis, cholinergic hypothesis, and excitotoxicity hypothesis. In the case of AD, CSF dysfunction is noticed even before cognitive decline. Activities of mTOR cause vascular irregularities in the brain decreasing cerebral blood flow which in turn sets up cognitive decline. The amyloid cascade hypothesis identifies the accumulation of Aβ plaques at different areas of CNS and related changes as the principal factor behind the development of AD
[97]. TAU hypothesis proposed that hyperphosphorylation of TAU leads to form neurofibrillary tangles preventing its regular role of supporting axonal microtubules and subsequently plays a critical role in neurodegeneration
[98]. Cholinergic hypothesis focuses on symptoms of cognitive decline and presents malfunctioning of cholinergic neurons as a pathophysiological factor towards initiation of AD
[99]. Excitotoxicity refers to the unprecedented death of nerve cells due to the overstimulation of certain amino acid receptors
[100]. A high concentration of glutamates activates N-methyl-d-aspartate and α-amino-3-hydroxy-5-methylisoxazole propionic acid receptors. As a result, voltage-gated calcium allows the entry of extracellular calcium into cells and thus a hindrance in neuronal energy metabolism leads to cell death.
 +1 credit
+1 credit