 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ji-Eun Chang | + 1914 word(s) | 1914 | 2021-09-09 05:22:47 | | | |
| 2 | Dean Liu | Meta information modification | 1914 | 2021-10-26 05:33:56 | | | | |
| 3 | Dean Liu | -11 word(s) | 1903 | 2021-10-27 04:42:31 | | |
Video Upload Options
Autophagy is a well-known intracellular elimination process, which provides degradation of damaged and dysfunctional organelles and proteins under highly-stressed conditions. This term came from the Greek word which represents ‘self-eating’.
1. Introduction
Inflammation is an adaptive response to tissue injury caused by infection, wound, or chemical irritation [1]. It is also known to be an essential process for the restoring of tissue functionality and homeostasis [2]. The relationship between inflammation and cancer has emerged as an important topic for numerous researchers for many years. In 1863, Rudolf Virchow suggested that the origin of cancer might be the chronic inflammation sites. This hypothesis was supported by further studies dealing with the association of chronic inflammatory diseases and enhanced cancer risks [3][4][5]. Now it has been established that inflammatory cells, as well as the chemokines and cytokines which they produce, strongly influence tumor development [6]. At the initiation stage of the neoplastic process, inflammatory cells act as effective cancer promoters. As the process progresses, they function to encourage cancer spread and metastasis. On the other hand, the inflammatory cells may also suppress tumor growth.
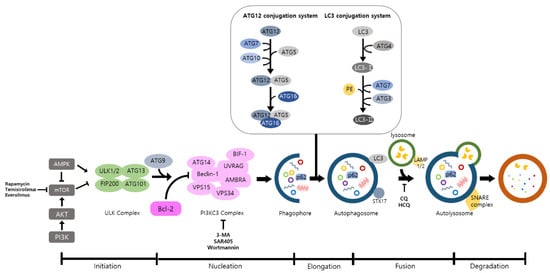
Autophagy is a well-known intracellular elimination process, which provides degradation of damaged and dysfunctional organelles and proteins under highly-stressed conditions. This term came from the Greek word which represents ‘self-eating’. Figure 1 shows the overall autophagic process and the inhibitors targeting each step. Several studies have shown that close communication between autophagy and apoptosis exists in determining the fate of cells under pathophysiological conditions [7][8]. It is well known that cells use autophagy as a survival mechanism to avoid cell death. However, when the stress increases to a level that exceeds the limits of cellular repair mechanisms, cells inhibit autophagy and initiate the apoptotic cascade [9]. The interaction between autophagy and apoptosis is very complex and entangled with necroptosis; autophagy can function either as an antiapoptotic mechanism or as a proapoptotic mechanism at various stages leading to cell death [10]. Recent research supports the idea that autophagy also plays a pivotal role in regulating inflammation [11]. Various signaling pathways which control tumor-associated inflammation regulate autophagy as well [12]. Inflammatory signals may either induce or inhibit the autophagy process, then autophagy influences tumor-associated inflammation such as induction or inhibition. In addition, autophagy may also regulate key inflammatory cytokines in tumors. Accordingly, autophagy leads to opposing consequences for the tumor since it works as both a tumor suppressor and a tumor promotor [13]. Figure 2 represents autophagy signaling pathways involved in cancer-associated inflammation. In this review, we discuss the relationship between autophagy and the key inflammatory processes in cancer.
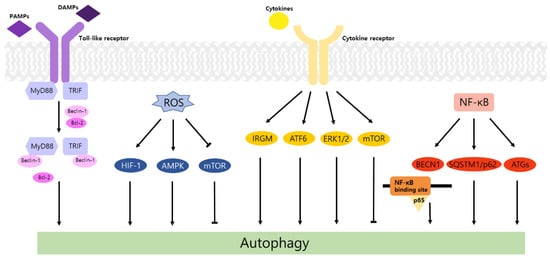
2. Autophagy and TLR Signaling in Cancer
Toll-like receptors (TLRs) are immune regulators that modulate inflammatory response. They specifically recognize damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) by the innate immune system [14]. Then activated TLRs stimulate diverse transcriptional pathways by the myeloid differentiation primary response gene 88 (MyD88)-dependent or MyD88-independent process. Among them, nuclear factor-κB (NF-κB), mitogen-activated protein kinase (MAPK), and interferon-regulatory factors (IRFs) pathways are associated with the transcription of cancer-related genes that regulate tumor progression, including inflammation, proliferation, angiogenesis, and metastasis. In particular, the NF-κB cascade induces the expression of cytokines, including tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), and IL-6, which may cause tumorigenic inflammation [15]. TLRs also regulate the autophagic activity via MyD88- and Toll/IL-1 receptor domain-containing adaptor-inducing interferon-β (TRIF)-dependent pathways. The conjugation of these proteins and Beclin-1 to TLR4 separates the Bcl-2/Beclin-1 complex and triggers the development of autophagosome [14].
In contrast, TLR4 enhances autophagy induction through beclin 1 (BECN1) ubiquitination by TNF receptor-associated factor 6 (TRAF6) promoting cell migration and invasion in various cancer cell lines [16]. Elevation of LC3-II levels and reduction in cell migration/invasion were detected in additional treatment with chloroquine (CQ), the lysosomal inhibitor. Moreover, the expression of IL-6, matrix metalloproteinase 2 (MMP2), and CC chemokine ligand 2 (CCL2) was augmented in A549 cells and was significantly higher in p62KO A549. These results indicate that TLR4 induces autophagy and cell migration/invasion, which is negatively controlled by p62.
Similarly, TLR9 induces autophagy and stimulates cell proliferation and invasion of HCC. TLR9, ATG, LC3B, and BECN1 were known to be expressed in human HCC cell line, Huh7. Additionally, epithelial mesenchymal transition (EMT) bioindicators, including vimentin and snail showed an increasing pattern as the TLR9 upregulated. Hydroxychloroquine (HCQ) is a suppressor of TLR9 and autophagy. Therefore, HCQ inhibits formation of autolysosome and affects progression of HCC. After treating with HCQ, TLR9 and EMT bioindicators were reduced, and cell viability and migration/invasion were also inhibited [17].
Taken together, TLRs stimulate the activation of autophagy and play a dual role in tumorigenesis. Different outcomes in tumor development are based on the type of TLR or cancer.
3. Regulation of Autophagy by ROS in Inflammation and Cancer
According to previous studies, ROS is known to stimulate autophagy. In breast and ovarian cancers, ROS enhances activity of autophagy and improves cancer cell survival [18]. Generated ROS induced autophagy by LC3 accumulation and increased the Beclin-1/Bcl-2 ratio [19]. In a recent study, when the ROS level is increased in prostate cancer, acidic vesicular organelles (AVO) and conversion of LC3-I to LC3-II hallmarks of autophagy are triggered [20]. In an oxygen and nutrient deficient environment, excess ROS increases the ratio of AMP/ATP, activates AMP-activated protein kinase (AMPK) [21], and induces formation of autophagosome via activation of UNC-51-like kinase (ULK) and phosphorylation of Beclin-1 [22]. ROS also induces autophagy by repressing the PI3K-Akt-mTOR pathway [23]. When cancer cells are under starvation conditions, H 2O 2 is generated, and cysteine protease ATG4 is restrained. This process elevates the formation of autophagy [24]. Recent studies demonstrated that the ROS level is increased in cancer cells and it stimulates autophagy. In colorectal and gastric cancers, beclin-1, the autophagy-related gene is highly expressed [25]. Autophagy impedes DNA mutations by preventing ROS accumulation through elimination of damaged mitochondria. Therefore, inhibition of autophagy via loss of Atg5 and Atg7 helps to initiate tumors by inducing accumulation of ROS and inflammation which is related to IL-1β, IL-18 [26].
Cancer cells show a high correlation with hypoxia due to their active metabolism. The translational factor, hypoxia-inducible factor (HIF), is the major mediator of hypoxia. The HIF-1 pathway encourages angiogenesis and provide oxygen to oxygen-deficient tissue [27]. In hypoxia, ROS is generated, and stabilized HIF-1α interacts with HIF-1β and binds to specific DNA sequence to make epigenetic changes [28]. This stimulates autophagy and promotes survival of cancer cells. Moreover, ROS upregulates cyclin mRNA which progresses the cell cycle from the G1 phase to the S phase. A low ROS level decreases cell proliferation and induces apoptosis since G1/S transition is restricted [29]. An excess of ROS, especially H 2O 2, reduces activity of MAP kinase phosphatase 3 (MKP3) and activates extracellular signal-regulated kinase (Erk)1/2 [30] to promote cell survival and adhesion-independent cell growth [31].
NADPH oxidase 2 induces ROS production and deactivates AMPK increasing colon cancer metastasis [32]. In addition, other important sources of ROS generation such as mitochondrial electron transport chain complexes [33], xanthine oxidase [34], and cyclooxygenase-2 [35] also play critical roles in the regulation of autophagy and the inflammatory process in cancer.
Taken together, ROS correlates with the whole inflammation process. Inflammatory factors influence activation of autophagy. ROS production is related to massive metabolic process in cancers and this leads to the autophagy process.
4. Autophagy and Inflammatory Cytokines in Cancer
IL-6 alleviates the induction of autophagy during tumor progression. In ovarian cancer cell NIH-OVCAR3, IL-6 blocked the accumulation of autophagosome and GFP-LC3 puncta at the area, which is deeply involved in migration activity. Furthermore, it was observed that cell migration was suppressed by IL-6 stimulation. These findings are associated with the expression of ARH-1 and AKT/mTOR/signal transducer and activator of transcription 3 (STAT3) pathway. ARH-1 acts as an autophagy inducer combining with Beclin-1 and activating the PI3KC3 complex. Data showed that IL-6 decreased the expression of ARH-1 and stimulated the phosphorylation of AKT, mTOR, and STAT3 and in turn, led to cell invasion [36].
Similarly, IL-8 secreted by cancer-associated fibroblasts (CAFs) also reduces autophagy and stimulates cell migration of ovarian cancer [37]. CAFs, major components of stromal cells, are included in the tumor microenvironment (TME) with immune cells and the extracellular matrix (ECM). The interplay between the tumor cell and TME, which is closely related to cytokines and autophagy, influences tumorigenesis [38]. Ovarian cancer-associated fibroblasts (OVCAFs) secreted a greater amount of IL-8 than ovarian normal-associated fibroblasts (OVNFs). Due to enhanced IL-8, LC3-II/LC3-I decreased, and p62 accumulation increased in human ovarian cancer cell lines. Moreover, cell migration was promoted in a wound healing assay.
Autophagy also regulates the level of cytokines in several cancers. Induced autophagy in CAFs can stimulate secretion of IL-6 and IL-8 followed by aggressive tumor development. In head and neck squamous cell carcinoma (HNSCC) cell lines, CAFs showed a potent autophagic activity compared to normal fibroblast (NF) and an increase in LC3-II under CQ treatment. Moreover, the outcomes of this study displayed a decrease in the density of IL-6 and IL-8 through knockdown of Beclin-1 and a reduction in cell migration and invasion of the tumor in siBECN, siATG7, or CQ-treated CAF-CM. The addition of IL-6 and IL-8 to siBECN promoted cell migration. Importantly, autophagy and cytokines such as IL-6 and IL-8 form a feedback loop during this process. IL-6 and IL-8 secreted by CAFs or HNSCC cells may stimulate CAF autophagy [39].
In summary, cytokines and autophagy represent protumor or antitumor effects under the unclear roles for each other, and the mechanisms have not been completely understood. The results discussed above showed the relation between the duality of autophagy in tumorigenesis and cytokines. Generally, it is known that IFN-γ, TNF-α, and IL-6 promote autophagy while IL-10 inhibits autophagy via diverse signaling cascades [40]. However, in this review we suggested that TNF-α and IL-6 may both promote and inhibit autophagy in certain cases. The action of autophagy on cytokine secretion has also been suggested. Autophagy increases the production of IL-6 and TNF-α in diverse cell types, which induces innate immune responses [40], and similar results were observed in cancer. Conversely, some studies showed that autophagy inhibited the expression of IL-6 [41] and TNF-α [42], and further studies are required to determine the relationship with tumorigenesis.
References
- Philip, M.; Rowley, D.A.; Schreiber, H. Inflammation as a tumor promoter in cancer induction. Semin. Cancer Biol. 2004, 14, 433–439.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Gurel, B.; Lucia, M.S.; Thompson, I.M.; Goodman, P.J.; Tangen, C.M.; Kristal, A.R.; Parnes, H.L.; Hoque, A.; Lippman, S.M.; Sutcliffe, S.; et al. Chronic inflammation in benign prostate tissue is associated with high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol. Biomark. Prev. 2014, 23, 847–856.
- McKay, C.J.; Glen, P.; McMillan, D.C. Chronic inflammation and pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 65–73.
- Michaud, D.S. Chronic inflammation and bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2007, 25, 260–268.
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867.
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349.
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010, 6, 891–900.
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A molecular switch node in the crosstalk between autophagy and apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083.
- Saleem, S. Apoptosis, Autophagy, Necrosis and Their Multi Galore Crosstalk in Neurodegeneration. Neuroscience 2021, 469, 162–174.
- Banskota, S.; Wang, H.; Kwon, Y.H.; Gautam, J.; Gurung, P.; Haq, S.; Hassan, F.M.N.; Bowdish, D.M.; Kim, J.A.; Carling, D.; et al. Salicylates ameliorate intestinal inflammation by activating macrophage AMPK. Inflamm. Bowel Dis. 2021, 27, 914–926.
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198.
- Choi, M.S.; Chae, Y.; Choi, J.W.; Chang, J. Potential Therapeutic Approaches through Modulating the Autophagy Process for Skin Barrier Dysfunction. Int. J. Mol. Sci. 2021, 22, 7869.
- Hayat, M.A. Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Academic Press: Cambridge, UK, 2017; Volume 12, ISBN 0128121475.
- Javaid, N.; Choi, S. Toll-like receptors from the perspective of cancer treatment. Cancers 2020, 12, 297.
- Kim, M.J.; Min, Y.; Im, J.S.; Son, J.; Lee, J.S.; Lee, K.Y. p62 is Negatively Implicated in the TRAF6-BECN1 Signaling Axis for Autophagy Activation and Cancer Progression by Toll-Like Receptor 4 (TLR4). Cells 2020, 9, 1142.
- Chen, M.-Y.; Yadav, V.K.; Chu, Y.C.; Ong, J.R.; Huang, T.-Y.; Lee, K.-F.; Lee, K.-H.; Yeh, C.-T.; Lee, W.-H. Hydroxychloroquine (HCQ) Modulates Autophagy and Oxidative DNA Damage Stress in Hepatocellular Carcinoma to Overcome Sorafenib Resistance via TLR9/SOD1/hsa-miR-30a-5p/Beclin-1 Axis. Cancers 2021, 13, 3227.
- Liu, W.; Glunde, K.; Bhujwalla, Z.M.; Raman, V.; Sharma, A.; Phang, J.M. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012, 72, 3677–3686.
- Yang, H.-L.; Liu, H.-W.; Shrestha, S.; Thiyagarajan, V.; Huang, H.-C.; Hseu, Y.-C. Antrodia salmonea induces apoptosis and enhances cytoprotective autophagy in colon cancer cells. Aging (Albany NY) 2021, 13, 15964–15989.
- Choi, H.D.; Kim, K.Y.; Park, K.I.; Kim, S.H.; Park, S.G.; Yu, S.N.; Kim, Y.W.; Kim, D.S.; Chung, K.T.; Ahn, S.C. Dual role of reactive oxygen species in autophagy and apoptosis induced by compound PN in prostate cancer cells. Mol. Cell. Toxicol. 2021, 17, 41–50.
- Pelosse, M.; Cottet-Rousselle, C.; Bidan, C.M.; Dupont, A.; Gupta, K.; Berger, I.; Schlattner, U. Synthetic energy sensor AMPfret deciphers adenylate-dependent AMPK activation mechanism. Nat. Commun. 2019, 10, 1–13.
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 1–15.
- Mickymaray, S.; Alfaiz, F.A.; Paramasivam, A.; Veeraraghavan, V.P.; Periadurai, N.D.; Surapaneni, K.M.; Niu, G. Rhaponticin suppresses osteosarcoma through the inhibition of PI3K-Akt-mTOR pathway. Saudi J. Biol. Sci. 2021, 28, 3641–3649.
- Abreu, S.; Kriegenburg, F.; Gómez-Sánchez, R.; Mari, M.; Sánchez-Wandelmer, J.; Skytte Rasmussen, M.; Soares Guimarães, R.; Zens, B.; Schuschnig, M.; Hardenberg, R.; et al. Conserved Atg8 recognition sites mediate Atg4 association with autophagosomal membranes and Atg8 deconjugation. EMBO Rep. 2017, 18, 765–780.
- Sun, Y.; Liu, J.H.; Jin, L.; Lin, S.M.; Yang, Y.; Sui, Y.X.; Shi, H. Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett. 2010, 294, 204–210.
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Schink, M.K.O.; Theodossiou, T.A.; Johansen, T.; Juhász, G.; Bilder, D.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420.
- Ebersole, J.L.; Novak, M.J.; Orraca, L.; Martinez-Gonzalez, J.; Kirakodu, S.; Chen, K.C.; Stromberg, A.; Gonzalez, O.A. Hypoxia-inducible transcription factors, HIF1A and HIF2A, increase in aging mucosal tissues. Immunology 2018, 154, 452–464.
- Palazon, A.; Goldrath, A.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528.
- Tai, L.; Huang, C.J.; Choo, K.B.; Cheong, S.K.; Kamarul, T. Oxidative stress down-regulates mir-20b-5p, mir-106a-5p and E2F1 expression to suppress the g1/s transition of the cell cycle in multipotent stromal cells. Int. J. Med. Sci. 2020, 17, 457–470.
- Chan, D.W.; Liu, V.W.S.; Tsao, G.S.W.; Yao, K.M.; Furukawa, T.; Chan, K.K.L.; Ngan, H.Y.S. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 2008, 29, 1742–1750.
- Chi, T.F.; Khoder-Agha, F.; Mennerich, D.; Kellokumpu, S.; Miinalainen, I.I.; Kietzmann, T.; Dimova, E.Y. Loss of USF2 promotes proliferation, migration and mitophagy in a redox-dependent manner. Redox Biol. 2020, 37, 1–9.
- Banskota, S.; Regmi, S.C.; Kim, J.A. NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in colon cancer cells through overexpression of MMP-7. Mol. Cancer 2015, 14, 1–14.
- Pramanik, K.C.; Boreddy, S.R.; Srivastava, S.K. Role of mitochondrial Electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS ONE 2011, 6, 20151.
- Huang, C.C.; Lee, C.C.; Lin, H.H.; Chen, M.C.; Lin, C.C.; Chang, J.Y. Autophagy-regulated ROS from xanthine oxidase acts as an early effector for triggering late mitochondria-dependent apoptosis in cathepsin S-targeted tumor cells. PLoS ONE 2015, 10, 1–20.
- Wang, Y.; Mandal, A.K.; Son, Y.O.K.; Pratheeshkumar, P.; Wise, J.T.F.; Wang, L.; Zhang, Z.; Shi, X.; Chen, Z. Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol. Appl. Pharmacol. 2018, 353, 23–30.
- Ferraresi, A.; Phadngam, S.; Morani, F.; Galetto, A.; Alabiso, O.; Chiorino, G.; Isidoro, C. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy. Mol. Carcinog. 2017, 56, 1164–1181.
- Thongchot, S.; Jamjuntra, P.; Therasakvichya, S.; Warnnissorn, M.; Ferraresi, A.; Thuwajit, P.; Isidoro, C.; Thuwajit, C. Interleukin-8 released by cancer-associated fibroblasts attenuates the autophagy and promotes the migration of ovarian cancer cells. Int. J. Oncol. 2021, 58, 1–14.
- Angrini, M.; Varthaman, A.; Cremer, I. Toll-like receptors (TLRs) in the tumor microenvironment (TME): A dragon-like weapon in a non-fantasy game of thrones. In Tumor Microenvironment; Springer: Cham, Switzerland, 2020; pp. 145–173.
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 2017, 77, 6679–6691.
- Mohan, S.; Gupta, D. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed. Pharmacother. 2018, 108, 1866–1878.
- Cotzomi-Ortega, I.; Rosas-Cruz, A.; Ramírez-Ramírez, D.; Reyes-Leyva, J.; Rodriguez-Sosa, M.; Aguilar-Alonso, P.; Maycotte, P. Autophagy inhibition induces the secretion of macrophage migration inhibitory factor (MIF) with autocrine and paracrine effects on the promotion of malignancy in breast cancer. Biology 2020, 9, 20.
- Pun, N.T.; Subedi, A.; Kim, M.J.; Park, P.H. Globular adiponectin causes tolerance to LPS-induced TNF-α expression via autophagy induction in RAW 264.7 macrophages: Involvement of SIRT1/FoxO3A axis. PLoS ONE 2015, 10, 1–22.

