 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Exequiel Medina | + 2017 word(s) | 2017 | 2021-09-28 05:37:05 | | | |
| 2 | Nora Tang | + 397 word(s) | 2414 | 2021-10-27 05:19:32 | | |
Video Upload Options
Although the Fox family of transcription factors has been described as monomers even in the presence of their cognate DNA, suggesting their full functionality without requiring oligomerization, members of the FoxP subfamily show both monomers and 3D-DS dimers (B). This novel ability in a well-known monomeric family has been largely attributed to a single replacement of a conserved proline by alanine (Pro39Ala) in the hinge region that connects helices H2 and H4 (B). Additionally, the ability to adopt intertwined dimers has been a focus of interest in terms of the possibility to bind different DNA loci within a given chromosome or even in physically mediating interchromosomal contacts, suggesting that the emergence of the 3D-DS could impact their mechanism of action and the complexity of the gene regulation networks in which they participate.
1. Introduction
An evolutionary advantage of oligomerization is that it allows the formation of large macromolecular complexes with additional functional sites in comparison to isolated monomers, all without increasing genome size [1][2]. Concomitantly, several protein structure databases convey that homooligomers are dominant over monomers [3][4], emphasizing the relevance of understanding the evolution of protein–protein interactions and their role in both structural and functional diversification in all organisms.
The emergence of oligomers in nature starting from ancestral monomers has been largely explained by the accumulation of random mutations on the surface of the monomers throughout evolution, which generates an optimized interface that enables protein–protein interactions [5][6]. These mutations allow the stabilization of hydrophobic interactions, salt bridges, and hydrogens bonds, which are the main interactions characterized as fundamental in enabling the stabilization of protein–protein interfaces during oligomer evolution [5][6]. However, three-dimensional domain swapping (3D-DS) has been proposed as an alternative and ancient mechanism for the evolution of protein oligomerization that does not require an optimized interaction surface [7]. In this mechanism, two (or more) protein chains exchange identical elements of their structures to form an intertwined oligomer [8] ( Figure 1 A).
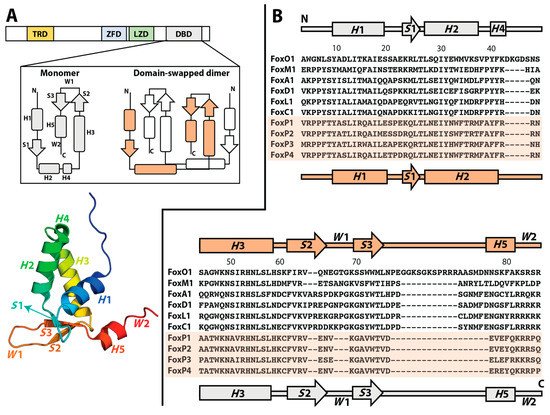
In order to reach a 3D-DS dimer, different intramolecular contacts that stabilize the monomeric structure must be broken and then recruited in an intermolecular fashion in the dimer. Since the residue pairs that participate in these quaternary contacts resemble those constituting monomeric contacts within a single polypeptide chain, there is no need to optimize a specific surface in an ancestral monomer to contact the partner subunit in the context of 3D-DS [7]. As a consequence, the only structural difference between a subunit in the 3D-DS dimer and its monomeric counterpart is the conformation of the hinge region, which connects the exchanging part in the intertwined dimer with the rest of its corresponding polypeptide chain [9] ( Figure 1 A).
Evidence of the evolutionary emergence of quaternary structures via 3D-DS in extant proteins is exemplary illustrated by histones. These are dimeric interleaved helical protein bundles where each monomer adopts a histone fold, common to all core eukaryotic histones and also present in archaeal histones and composed by two contiguous helix–strand–helix motifs connected as a result of a tandem duplication [10]. A sequence-based search for distant homologs employing Hidden Markov Models led to the identification of other protein folds also containing tandem duplications of these helix–strand–helix motifs, such as the C-domain of AAA+ ATPase proteins [11]. However, the C-domain is topologically different to the histone dimer in that it involves the association of the two consecutive motifs in a single polypeptide chain to constitute a four-helix bundle instead of protein–protein interactions between two monomers [11], which is a description that is consistent with the differences between isolated monomers and 3D-DS dimers.
2. Molecular Evolution towards 3D-DS in Human FoxP Transcription Factors
While 3D-DS offers a simple evolutionary pathway to the emergence of oligomerization that does not require a specialized protein–protein interface, it is accompanied by the biophysical intricacy of requiring the breakage of many intramolecular contacts through at least partial protein unfolding to form an intertwined dimer [12]. Since the first description of 3D-DS in diphtheria toxin [7], seminal cases such as cyanovirin [13] and the yeast cell cycle controller p13suc1 [14] have shown that dimerization via 3D-DS is dramatically accelerated when these proteins are exposed to structural perturbations, i.e., pH and temperature changes or the addition of chemical denaturing agents, which is followed by restoring physiological conditions while employing high protein concentrations [15]. These observations demonstrate that reaching the unfolded state is a main limiting step to adopting the dimer for most studied cases. Moreover, the need of protein unfolding to speed up 3D-DS obscures the elucidation of the physiological significance of these intertwined oligomers [12].
Strikingly, a recent example of 3D-DS under physiological conditions has been observed in the FoxP subfamily of transcription factors. Fox proteins are present in yeast and metazoans, and several evolutionary analyses have strongly suggested that these proteins were present in the ancestor of all eukaryotes [16][17]. Their absence in plants suggests that their origin is linked to a clade of unicellular organisms that gave rise to both the fungal and animal lineages [18]. Moreover, evolutionary analyses combined with gene expression studies in the ancient invertebrate chordate amphioxus have demonstrated that a cluster of four Fox genes with sequential and coordinated endo-mesodermal tissue expression has been present since basal Bilateria and maintained in several lineages during animal evolution for more than 500 million years [19].
According to most solved Fox structures to date, their DBDs exist as monomers with helix H 3 contacting the major groove of DNA [20][21][22][23][24]. While a canonical sequence RYAAAYA located on promoter regions has been defined as the target for Fox DBDs, phylogenetic and functional DNA-binding studies among the family found that some DBDs can bind to alternative DNA sequence motifs in addition to the canonical ones [25]. These results showed that changes in DNA-binding specificity across the Fox family were not explained by changes in DNA-contacting amino acids that define the specificity for canonical DNA binding sites. Moreover, some Fox proteins can specifically bind two sequence motifs. To explain these results, Nakagawa et al. [25] proposed that the DBD could adopt an alternative conformation with respect to the one observed in solved structures, allowing the recognition of additional DNA binding motifs. These insights suggest that structural heterogeneity may play a crucial role in the function of Fox proteins.
Regardless of the sequence and structure similarity between Fox proteins, they are differentially involved in several regulatory networks related to proliferation, differentiation, angiogenesis, apoptosis, and cell cycle progression [26]. Moreover, Fox subfamilies such as FoxC, FoxM, FoxP, and FoxA have been closely related to cancer [26][27]. Indeed, FoxP members have been described as oncogenes or tumor suppressors depending on cellular contexts [28], revealing the complexity of transcriptional networks in which these proteins are involved.
3. Biophysically Dissecting the Evolutionary Strategies of FoxP Proteins to Overcome the Thermodynamic Limitations of 3D-DS
As mentioned above, 3D-DS emerged in the FoxP subfamily as an evolutionary novelty to promote protein association. However, most of the studied 3D-DS models to date conclude that the acquisition of the intertwined dimer (or oligomer) is kinetically limited by protein unfolding [13][14][15][29]. Although this behavior has been widely observed in canonical examples of 3D-DS [29][30][31], the human FoxP subfamily has shown different kinetic and thermodynamic properties when compared with such models [32][33][34][35][36].
One of the main hypotheses that explains the properties shown by FoxP1 is a decrease in protein stability. Specifically, the decrease in the energy barrier that imposes the loss of native contacts in FoxP1 could be compensated by the intermolecular stabilization of the structure once the dimer is adopted [37][38][39][40]. In this line, NMR analysis of the monomeric and dimeric states of FoxP1 indicated that helices H 2 and H 3, and the wing W 1 are notably flexible compared with the other secondary structures in the chain [34]. Thus, stability changes could promote at least local unfolding. In addition, these analyses determined that the dimer is more flexible throughout its backbone when compared with the monomer, giving preliminary clues about the role of structural flexibility in the stability and propensity of 3D-DS.
In order to gain more insights regarding local structural changes in FoxP1 upon 3D-DS, hydrogen–deuterium exchange coupled with mass spectrometry (HDXMS) was used. This experimental strategy ascertains the solvent accessibility and/or flexibility of local regions of a given protein through its incubation in a deuterated buffer, such that the extent of deuteron incorporation from the solvent into the backbone amides of a protein acts as a mass probe, which is followed by its proteolytic digestion and final analysis by mass spectrometry to monitor these changes over local protein regions ( Figure 2 ) [41][42]. In the case of FoxP1, HDXMS experiments on the wild-type protein under physiological and mild-denaturing conditions described regions with high flexibility (as determined by their higher deuteron incorporation when compared to other local regions within the protein), such as helices H 1 and H 5, and beta strand S 1 ( Figure 2 ), suggesting the relevance of these localized regions for both the monomeric intermediate acquisition as well as favoring the dimerization of FoxP1 [35]. Moreover, the comparison between FoxP1 and the mutant Ala39Pro showed a stabilization of the region H 1- S 1 that could be a relevant factor to modulate 3D-DS in FoxP proteins, further strengthening that local rather than global structural perturbations suffice to facilitate their dimerization.
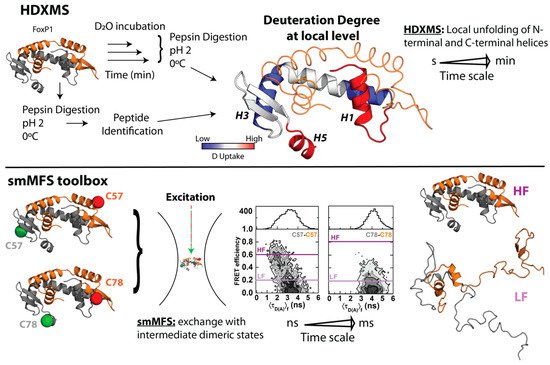
Altogether, these experiments represent the first indications of an evolutive strategy in this subfamily to overcome the thermodynamic limitations of 3D-DS. A decrease in protein stability and an increase in local structural flexibility seemed to be correlated with the dimerization (and dissociation) properties of FoxP proteins. However, none of those approaches explain how these structural features relate with the mechanism by which FoxP associates via 3D-DS.
4. Evolution Pathway inside FoxP Subfamily and Their Impact in Functionality: From Homodimers to Heterodimers and Beyond
While FoxP1 is the most biophysically characterized member in the subfamily, these observations could be described as general features for all FoxP members given that the DBDs of these transcription factors exhibit a high degree of sequence identity (75–92%).
An evolutionary pathway forming a secondary interface to enhance intersubunit contacts in a 3D-DS dimer was suggested as the primitive mechanism to form stable intertwined dimers in IFN-y, IL-5, and βb2-crystallin [7]. Since those proteins do not have a monomeric structure, comparing their intertwined dimer with closely monomeric homologs structures showed that the interdomain interface of the dimer resembles the monomeric interdomain interface. Therefore, domain swapping could be the first step in the evolution of these dimers, which is followed by the formation of a secondary interface [7]. Considering the aforementioned higher dissociation constants and higher sequence identity between FoxP1, FoxP2, and FoxP4 when compared with FoxP3, the FoxP family constitutes an excellent model to dissect the mutations accumulated throughout evolution that provide primary and secondary interfaces for the emergence of 3D-DS and its specialization into obligated dimers, and to examine the different dimerization properties exhibited by these transcription factors in the context of their direct impacts on functional diversification.
Several computational studies have indicated that 3D-DS depends primarily on the monomer’s topology [37][39][43] and the structural characteristics of the hinge loop [14][44][45]. Experimental studies on covalently fused immunoglobulin domains with varying degrees of sequence identity from the muscle protein titin showed that their propensity of aggregation into dimers is high when the sequence identity is above 70% [46], such that low sequence identity is necessary to avoid misfolding. Similar studies at the single-molecule level and accompanied by native-centric molecular dynamics demonstrated that the interactions underlying misfolding in these domains are the result of sequence-specific 3D-DS [47]. Building upon these results, molecular dynamics simulations using force fields that take into account the sequence-dependence of protein interactions demonstrated that the reduction of the sequence identity between covalently linked multidomains decreased the formation of 3D-DS contacts, and that these contacts compete with other strong hydrophobic self-recognition contacts, leading to non-3D-DS misfolding in a balance that can be modulated by point mutations [48].
These insights suggest the possibility of heteroassociation via 3D-DS in the FoxP subfamily due to their high sequence identity. Although some studies showed the ability to heterodimerize between FoxP members [49][50], this process has not been biophysically explored, opening an interesting question about the functional and structural role of FoxP1 in increasing the association ability of the other FoxP members.
References
- Blundell, T.L.; Srinivasan, N. Symmetry, Stability, and Dynamics of Multidomain and Multicomponent Protein Systems. Proc. Natl. Acad. Sci. USA 1996, 93, 14243–14248.
- Goodsell, D.S.; Olson, A.J. Structural Symmetry and Protein Function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153.
- Marianayagam, N.J.; Sunde, M.; Matthews, J.M. The Power of Two: Protein Dimerization in Biology. Trends Biochem. Sci. 2004, 29, 618–625.
- Marsh, J.A.; Teichmann, S.A. Structure, Dynamics, Assembly, and Evolution of Protein Complexes. Annu. Rev. Biochem. 2015, 84, 551–575.
- Jones, S.; Thornton, J.M. Protein-Protein Interactions: A Review of Protein Dimer Structures. Prog. Biophys. Mol. Biol. 1995, 63, 31–65.
- Gwyther, R.E.A.; Jones, D.D.; Worthy, H.L. Better Together: Building Protein Oligomers Naturally and by Design. Biochem. Soc. Trans. 2019, 47, 1773–1780.
- Bennett, M.J.; Choe, S.; Eisenberg, D. Domain Swapping: Entangling Alliances between Proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 3127–3131.
- Liu, Y.; Eisenberg, D. 3D Domain Swapping: As Domains Continue to Swap. Protein Sci. 2002, 11, 1285–1299.
- Bennett, M.J.; Schlunegger, M.P.; Eisenberg, D. 3D Domain Swapping: A Mechanism for Oligomer Assembly. Protein Sci. 1995, 4, 2455–2468.
- Arents, G.; Moudrianakis, E.N. The Histone Fold: A Ubiquitous Architectural Motif Utilized in DNA Compaction and Protein Dimerization. Proc. Natl. Acad. Sci. USA 1995, 92, 11170–11174.
- Alva, V.; Ammelburg, M.; Söding, J.; Lupas, A.N. On the Origin of the Histone Fold. BMC Struct. Biol. 2007, 7, 17.
- Rousseau, F.; Schymkowitz, J.; Itzhaki, L.S. Implications of 3D Domain Swapping for Protein Folding, Misfolding and Function. Adv. Exp. Med. Biol. 2012, 747, 137–152.
- Liu, L.; Byeon, I.J.L.; Bahar, I.; Gronenborn, A.M. Domain Swapping Proceeds via Complete Unfolding: A 19F- and 1H-NMR Study of the Cyanovirin-N Protein. J. Am. Chem. Soc. 2012, 134, 4229–4235.
- Rousseau, F.; Schymkowitz, J.W.; Wilkinson, H.R.; Itzhaki, L.S. Three-Dimensional Domain Swapping in p13suc1 Occurs in the Unfolded State and Is Controlled by Conserved Proline Residues. Proc. Natl. Acad. Sci. USA 2001, 98, 5596–5601.
- Rousseau, F.; Schymkowitz, J.W.H.; Itzhaki, L.S. The Unfolding Story of Three-Dimensional Domain Swapping. Structure 2003, 11, 243–251.
- Mazet, F.; Yu, J.K.; Liberles, D.A.; Holland, L.Z.; Shimeld, S.M. Phylogenetic Relationships of the Fox (Forkhead) Gene Family in the Bilateria. Gene 2003, 316, 79–89.
- Pascual-Carreras, E.; Herrera-Úbeda, C.; Rosselló, M.; Garcia-Fernandez, J.; Saló, E.; Adell, T. Analysis of Fox Genes in Schmidtea Mediterranea Reveals New Families and a Conserved Role of Smed-foxO in Controlling Cell Death. Sci. Rep. 2021, 11, 2947.
- Baldauf, S.L. A Search for the Origins of Animals and Fungi: Comparing and Combining Molecular Data. Am. Nat. 1999, 154, S178–S188.
- Mazet, F.; Amemiya, C.T.; Shimeld, S.M. An Ancient Fox Gene Cluster in Bilaterian Animals. Curr. Biol. 2006, 16, R314–R316.
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-Crystal Structure of the HNF-3/fork Head DNA-Recognition Motif Resembles Histone H5. Nature 1993, 364, 412–420.
- Boura, E.; Rezabkova, L.; Brynda, J.; Obsilova, V.; Obsil, T. Structure of the human FOXO4-DBD-DNA complex at 1.9 Å resolution reveals new details of FOXO binding to the DNA. Acta. Crystallogr. D Biol. Crystallogr. 2010, 66, 1351–1357.
- Li, J.; Dai, S.; Chen, X.; Liang, X.; Qu, L.; Jiang, L.; Guo, M.; Zhou, Z.; Wei, H.; Zhang, H.; et al. Mechanism of forkhead transcription factors binding to a novel palindromic DNA site. Nucleic Acids Res 2021, 49, 3573–3583.
- Li, S.; Pradhan, L.; Ashur, S.; Joshi, A.; Nam, H.-J. Crystal Structure of FOXC2 in Complex with DNA Target. ACS Omega 2019, 4, 10906–10914.
- Chen, X.; Wei, H.; Li, J.; Liang, X.; Dai, S.; Jiang, L.; Guo, M.; Qu, L.; Chen, Z.; Chen, L.; et al. Structural Basis for DNA Recognition by FOXC2. Nucleic Acids Res. 2019, 47, 3752–3764.
- Nakagawa, S.; Gisselbrecht, S.S.; Rogers, J.M.; Hartl, D.L.; Bulyk, M.L. DNA-Binding Specificity Changes in the Evolution of Forkhead Transcription Factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12349–12354.
- Lam, E.W.-F.; Brosens, J.J.; Gomes, A.R.; Koo, C.-Y. Forkhead Box Proteins: Tuning Forks for Transcriptional Harmony. Nat. Rev. Cancer 2013, 13, 482–495.
- Bach, D.-H.; Long, N.P.; Luu, T.-T.-T.; Anh, N.H.; Kwon, S.W.; Lee, S.K. The Dominant Role of Forkhead Box Proteins in Cancer. Int. J. Mol. Sci. 2018, 19, 3279.
- Kim, J.-H.; Hwang, J.; Jung, J.H.; Lee, H.-J.; Lee, D.Y.; Kim, S.-H. Molecular Networks of FOXP Family: Dual Biologic Functions, Interplay with Other Molecules and Clinical Implications in Cancer Progression. Mol. Cancer 2019, 18, 180.
- Liu, Z.; Huang, Y. Evidences for the Unfolding Mechanism of Three-Dimensional Domain Swapping. Protein Sci. 2013, 22, 280–286.
- Janowski, R.; Kozak, M.; Jankowska, E.; Grzonka, Z.; Grubb, A.; Abrahamson, M.; Jaskolski, M. Human Cystatin C, an Amyloidogenic Protein, Dimerizes through Three-Dimensional Domain Swapping. Nat. Struct. Biol. 2001, 8, 316–320.
- Hafner-Bratkovic, I.; Bester, R.; Pristovsek, P.; Gaedtke, L.; Veranic, P.; Gaspersic, J.; Mancek-Keber, M.; Avbelj, M.; Polymenidou, M.; Julius, C.; et al. Globular Domain of the Prion Protein Needs to Be Unlocked by Domain Swapping to Support Prion Protein Conversion. J. Biol. Chem. 2011, 286, 12149–12156.
- Stroud, J.C.; Wu, Y.; Bates, D.L.; Han, A.; Nowick, K.; Paabo, S.; Tong, H.; Chen, L. Structure of the Forkhead Domain of FOXP2 Bound to DNA. Structure 2006, 14, 159–166.
- Bandukwala, H.S.; Wu, Y.; Feuerer, M.; Chen, Y.; Barboza, B.; Ghosh, S.; Stroud, J.C.; Benoist, C.; Mathis, D.; Rao, A.; et al. Structure of a Domain-Swapped FOXP3 Dimer on DNA and Its Function in Regulatory T Cells. Immunity 2011, 34, 479–491.
- Chu, Y.P.; Chang, C.H.; Shiu, J.H.; Chang, Y.T.; Chen, C.Y.; Chuang, W.J. Solution Structure and Backbone Dynamics of the DNA-Binding Domain of FOXP1: Insight into Its Domain Swapping and DNA Binding. Protein Sci. 2011, 20, 908–924.
- Medina, E.; Córdova, C.; Villalobos, P.; Reyes, J.; Komives, E.A.; Ramírez-Sarmiento, C.A.; Babul, J. Three-Dimensional Domain Swapping Changes the Folding Mechanism of the Forkhead Domain of FoxP1. Biophys. J. 2016, 110, 2349–2360.
- Medina, E.; Villalobos, P.; Coñuecar, R.; Ramírez-Sarmiento, C.A.; Babul, J. The Protonation State of an Evolutionarily Conserved Histidine Modulates Domain Swapping Stability of FoxP1. Sci. Rep. 2019, 9, 5441.
- Ding, F.; Prutzman, K.C.; Campbell, S.L.; Dokholyan, N.V. Topological Determinants of Protein Domain Swapping. Structure 2006, 14, 5–14.
- Garcia-Pino, A.; Martinez-Rodriguez, S.; Wahni, K.; Wyns, L.; Loris, R.; Messens, J. Coupling of Domain Swapping to Kinetic Stability in a Thioredoxin Mutant. J. Mol. Biol. 2009, 385, 1590–1599.
- Yang, S.; Cho, S.S.; Levy, Y.; Cheung, M.S.; Levine, H.; Wolynes, P.G.; Onuchic, J.N. Domain Swapping Is a Consequence of Minimal Frustration. Proc. Natl. Acad. Sci. USA 2004, 101, 13786–13791.
- Cho, S.S.; Levy, Y.; Onuchic, J.N.; Wolynes, P.G. Overcoming Residual Frustration in Domain-Swapping: The Roles of Disulfide Bonds in Dimerization and Aggregation. Phys. Biol. 2005, 2, S44–S55.
- Balasubramaniam, D.; Komives, E.A. Hydrogen-Exchange Mass Spectrometry for the Study of Intrinsic Disorder in Proteins. Biochim. Biophys. Acta 2013, 1834, 1202–1209.
- Ramirez-Sarmiento, C.A.; Komives, E.A. Hydrogen-Deuterium Exchange Mass Spectrometry Reveals Folding and Allostery in Protein-Protein Interactions. Methods 2018, 144, 43–52.
- Mascarenhas, N.M.; Gosavi, S. Protein Domain-Swapping Can Be a Consequence of Functional Residues. J. Phys. Chem. B 2016, 120, 6929–6938.
- Picone, D.; Di Fiore, A.; Ercole, C.; Franzese, M.; Sica, F.; Tomaselli, S.; Mazzarella, L. The Role of the Hinge Loop in Domain Swapping. The Special Case of Bovine Seminal Ribonuclease. J. Biol. Chem. 2005, 280, 13771–13778.
- Shingate, P.; Sowdhamini, R. Analysis of Domain-Swapped Oligomers Reveals Local Sequence Preferences and Structural Imprints at the Linker Regions and Swapped Interfaces. PLoS ONE 2012, 7, e39305.
- Wright, C.F.; Teichmann, S.A.; Clarke, J.; Dobson, C.M. The Importance of Sequence Diversity in the Aggregation and Evolution of Proteins. Nature 2005, 438, 878–881.
- Borgia, M.B.; Borgia, A.; Best, R.B.; Steward, A.; Nettels, D.; Wunderlich, B.; Schuler, B.; Clarke, J. Single-Molecule Fluorescence Reveals Sequence-Specific Misfolding in Multidomain Proteins. Nature 2011, 474, 662–665.
- Zheng, W.; Schafer, N.P.; Wolynes, P.G. Frustration in the Energy Landscapes of Multidomain Protein Misfolding. Proc. Natl. Acad. Sci. USA 2013, 110, 1680–1685.
- Li, S.; Weidenfeld, J.; Morrisey, E.E. Transcriptional and DNA Binding Activity of the Foxp1/2/4 Family Is Modulated by Heterotypic and Homotypic Protein Interactions. Mol. Cell. Biol. 2004, 24, 809–822.
- Wang, B.; Lin, D.; Li, C.; Tucker, P. Multiple Domains Define the Expression and Regulatory Properties of Foxp1 Forkhead Transcriptional Repressors. J. Biol. Chem. 2003, 278, 24259–24268.

