 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura Nuñez-Gonzalez | + 4752 word(s) | 4752 | 2021-10-25 05:15:43 | | | |
| 2 | Vivi Li | + 129 word(s) | 4881 | 2021-10-25 11:58:10 | | | | |
| 3 | Vivi Li | Meta information modification | 4881 | 2021-10-29 08:36:31 | | |
Video Upload Options
Gitelman and Bartter syndromes are rare inherited diseases that belong to the category of renal tubulopathies. The genes associated with these pathologies encode electrolyte transport proteins located in the nephron, particularly in the Distal Convoluted Tubule and Ascending Loop of Henle. Therefore, both syndromes are characterized by alterations in the secretion and reabsorption processes that occur in these regions. Patients suffer from deficiencies in the concentration of electrolytes in the blood and urine, which leads to different systemic consequences related to these salt-wasting processes. The main clinical features of both syndromes are hypokalemia, hypochloremia, metabolic alkalosis, hyperreninemia and hyperaldosteronism. Despite having a different molecular etiology, Gitelman and Bartter syndromes share a relevant number of clinical symptoms, and they have similar therapeutic approaches. The main basis of their treatment consists of electrolytes supplements accompanied by dietary changes. Specifically for Bartter syndrome, the use of non-steroidal anti-inflammatory drugs is also strongly supported.
1. Introduction
| Bartter Subtype | OMIM | Inheritance | Causative Gene | Related Protein | UniProt Code |
|---|---|---|---|---|---|
| Type 1 | 601,678 | AR | SLC12A1 | NKCC2 (Solute carrier family 12 member 1) |
Q13621 |
| Type 2 | 600,359 | AR | KCNJ1 | ROMK (ATP-sensitive inward rectifier potassium channel 1) |
P48048 |
| Type 3 | 607,364 | AR | CLCNKB | CLCNKB (Chloride channel protein ClC-Kb) |
P51801 |
| Type 4a | 602,522 | AR | BSND | BSND (Barttin) |
Q8WZ55 |
| Type 4b | 613,090 | AR DR | CLCNKB + CLCNKA |
CLCNKB (Chloride channel protein ClC-Kb) + CLCNKA (Chloride channel protein ClC-Ka) |
P51801 + P51800 |
| Type 5 | 300,470 | XLR | MAGED2 | MAGED2 (Melanoma-associated antigen D2) |
Q9UNF1 |
2. Molecular Basis and Clinical Features of the Diseases
2.1. Molecular Basis of Gitelman Syndrome and Clinical Consequences
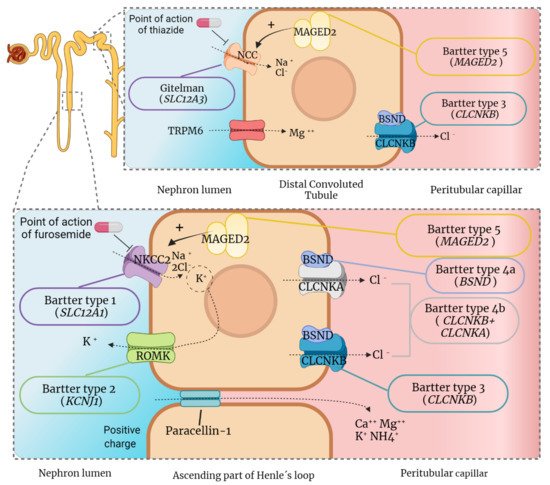
2.2. Molecular Basis of Bartter Syndrome and Clinical Consequences
-
Bartter syndrome types 1 and 2
-
Bartter syndrome types 3 and 4
2.3. Long-Term Outcomes in GS and BS
3. Therapeutic Approaches
3.1. Current Pharmacological Treatments
3.1.1. Oral Salt Supplementation
3.1.2. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
3.1.3. Potassium Sparing Diuretics
3.1.4. Renin-Angiotensin-Aldosterone System Inhibitors
3.1.5. Growth Hormone (GH)
3.1.6. Overview of the Effectiveness of These Treatments
| Therapeutic Approaches | Gitelman Syndrome | Bartter Syndrome |
|---|---|---|
| Supplemental electrolyte drugs | Mandatory, especially with magnesium loss | Mandatory |
| NSAIDs | Possible | Indomethacin as principal treatment in BS |
| Potassium Sparing Diuretics | Possible, but not recommendable | Possible, but not recommendable |
| Inhibitors of RAAS axis | Poorly described, but possible | Possible, especially with nephrotic damage from NSAIDs |
| Growth Hormone | Possible, poor evidence of efficacy | Possible, poor evidence of efficacy |
3.2. Future Therapeutic Perspectives
References
- Arakawa, H.; Kubo, H.; Washio, I.; Staub, A.Y.; Nedachi, S.; Ishiguro, N.; Nakanishi, T.; Tamai, I. Rat Kidney Slices for Evaluation of Apical Membrane Transporters in Proximal Tubular Cells. J. Pharm. Sci. 2019, 108, 2798–2804.
- Downie, M.L.; Lopez Garcia, S.C.; Kleta, R.; Bockenhauer, D. Inherited Tubulopathies of the Kidney. Clin. J. Am. Soc. Nephrol. 2020, 16, CJN14481119.
- Mitchell, J.E.; Pomeroy, C.; Seppala, M.; Huber, M. Pseudo-Bartter’s syndrome, diuretic abuse, idiopathic edema, and eating disorders. Int. J. Eat. Disord. 1988, 7, 225–237.
- Bartter and Gitelman Syndromes—UpToDate. Available online: https://www.uptodate.com/contents/inherited-hypokalemic-salt-losing-tubulopathies-pathophysiology-and-overview-of-clinical-manifestations (accessed on 14 May 2021).
- Hureaux, M.; Ashton, E.; Dahan, K.; Houillier, P.; Blanchard, A.; Cormier, C.; Koumakis, E.; Iancu, D.; Belge, H.; Hilbert, P.; et al. High-throughput sequencing contributes to the diagnosis of tubulopathies and familial hypercalcemia hypocalciuria in adults. Kidney Int. 2019, 96, 1408–1416.
- Bao, M.; Cai, J.; Yang, X.; Ma, W. Genetic screening for Bartter syndrome and Gitelman syndrome pathogenic genes among individuals with hypertension and hypokalemia. Clin. Exp. Hypertens. 2019, 41, 381–388.
- Orphanet: Bartter Syndrome. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=112 (accessed on 26 May 2021).
- Frederic, C. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am. J. Med. 1962, 33, 811–828.
- Simon, D.B.; Karet, F.E.; Hamdan, J.M.; Di Pietro, A.; Sanjad, S.A.; Lifton, R.P. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2CI cotransporter NKCC2. Nat. Genet. 1996, 13, 183–188.
- Simon, D.B.; Karet, F.E.; Rodriguez-Soriano, J.; Hamdan, J.H.; DiPietro, A.; Trachtman, H.; Sanjad, S.A.; Lifton, R.P. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet. 1996, 14, 152–156.
- Simon, D.B.; Bindra, R.S.; Mansfield, T.A.; Nelson-Williams, C.; Mendonca, E.; Stone, R.; Schurman, S.; Nayir, A.; Alpay, H.; Bakkaloglu, A.; et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 1997, 17, 171–178.
- Birkenhäger, R.; Otto, E.; Schürmann, M.J.; Vollmer, M.; Ruf, E.M.; Maier-Lutz, I.; Beekmann, F.; Fekete, A.; Omran, H.; Feldmann, D.; et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 2001, 29, 310–314.
- Laghmani, K.; Beck, B.B.; Yang, S.-S.; Seaayfan, E.; Wenzel, A.; Reusch, B.; Vitzthum, H.; Priem, D.; Demaretz, S.; Bergmann, K.; et al. Polyhydramnios, Transient Antenatal Bartter’s Syndrome, and MAGED2 Mutations. N. Engl. J. Med. 2016, 374, 1853–1863.
- Schlingmann, K.P.; Konrad, M.; Jeck, N.; Waldegger, P.; Reinalter, S.C.; Holder, M.; Seyberth, H.W.; Waldegger, S. Salt Wasting and Deafness Resulting from Mutations in Two Chloride Channels. N. Engl. J. Med. 2004, 350, 1314–1319.
- Bichet, D.G.; Fujiwara, T.M. Reabsorption of Sodium Chloride—Lessons from the Chloride Channels. N. Engl. J. Med. 2004, 350, 1281–1283.
- Gitelman, H.J.; Graham, J.B.; Welt, L.G. A familial disorder characterized by hypokalemia and hypomagnesemia. Ann. N. Y. Acad. Sci. 1969, 162, 856–864.
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996, 12, 24–30.
- Punzi, L.; Calo, L.; Schiavon, F.; Pianon, M.; Rosada, M.; Todesco, S. Chondrocalcinosis is a feature of Gitelman’s variant of Bartter’s syndrome: A new look at the hypomagnesemia associated with calcium pyrophosphate dihydrate crystal deposition disease. Rev. Rhum. 1998, 65, 571–574.
- Kleta, R.; Bockenhauer, D. Salt-losing tubulopathies in children: What’s new, what’s controversial? J. Am. Soc. Nephrol. 2018, 29, 727–739.
- Besouw, M.T.P.; Kleta, R.; Bockenhauer, D. Bartter and Gitelman syndromes: Questions of class. Pediatr. Nephrol. 2020, 35, 1815–1824.
- Nijenhuis, T.; Vallon, V.; Van Der Kemp, A.W.C.M.; Loffing, J.; Hoenderop, J.G.J.; Bindels, R.J.M. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J. Clin. Investig. 2005, 115, 1651–1658.
- Blanchard, A.; Bockenhauer, D.; Bolignano, D.; Calò, L.A.; Cosyns, E.; Devuyst, O.; Ellison, D.H.; Karet Frankl, F.E.; Knoers, N.V.A.M.; Konrad, M.; et al. Gitelman syndrome: Consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. In Proceedings of the Kidney International; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 91, pp. 24–33.
- Seys, E.; Andrini, O.; Keck, M.; Mansour-Hendili, L.; Courand, P.Y.; Simian, C.; Deschenes, G.; Kwon, T.; Bertholet-Thomas, A.; Bobrie, G.; et al. Clinical and genetic spectrum of bartter syndrome type 3. J. Am. Soc. Nephrol. 2017, 28, 2540–2552.
- Dai, L.J.; Ritchie, G.; Kerstan, D.; Kang, H.S.; Cole, D.E.C.; Quamme, G.A. Magnesium transport in the renal distal convoluted tubule. Physiol. Rev. 2001, 81, 51–84.
- Franken, G.A.C.; Adella, A.; Bindels, R.J.M.; de Baaij, J.H.F. Mechanisms coupling sodium and magnesium reabsorption in the distal convoluted tubule of the kidney. Acta Physiol. 2021, 231, e13528.
- Maeoka, Y.; McCormick, J.A. NaCl cotransporter activity and Mg2+ handling by the distal convoluted tubule. Am. J. Physiol.—Ren. Physiol. 2020, 319, F1043–F1053.
- Ea, H.K.; Blanchard, A.; Dougados, M.; Roux, C. Chondrocalcinosis secondary to hypomagnesemia in Gitelman’s syndrome. J. Rheumatol. 2005, 32, 1840–1842.
- Leone, F.A.; Rezende, L.A.; Ciancaglini, P.; Pizauro, J.M. Allosteric modulation of pyrophosphatase activity of rat osseous plate alkaline phosphatase by magnesium ions. Int. J. Biochem. Cell Biol. 1998, 30, 89–97.
- Calò, L.; Punzi, L.; Semplicini, A. Hypomagnesemia and chondrocalcinosis in Bartter’s and Gitelman’s syndrome: Review of the pathogenetic mechanisms. Am. J. Nephrol. 2000, 20, 347–350.
- Pollak, M.R.; Friedman, D.J. The genetic architecture of kidney disease. Clin. J. Am. Soc. Nephrol. 2020, 15, 268–275.
- Stewart, D.; Iancu, D.; Ashton, E.; Courtney, A.E.; Connor, A.; Walsh, S.B. Transplantation of a Gitelman Syndrome Kidney Ameliorates Hypertension: A Case Report. Am. J. Kidney Dis. 2019, 73, 421–424.
- Ji, W.; Foo, J.N.; O’Roak, B.J.; Zhao, H.; Larson, M.G.; Simon, D.B.; Newton-Cheh, C.; State, M.W.; Levy, D.; Lifton, R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008, 40, 592–599.
- Monette, M.Y.; Rinehart, J.; Lifton, R.P.; Forbush, B. Rare mutations in the human NA-K-CL cotransporter (NKCC2) associated with lower blood pressure exhibit impaired processing and transport function. Am. J. Physiol.—Ren. Physiol. 2011, 300, 840–847.
- Balavoine, A.S.; Bataille, P.; Vanhille, P.; Azar, R.; Noël, C.; Asseman, P.; Soudan, B.; Wémeau, J.L.; Vantyghem, M.C. Phenotype-genotype correlation and follow-up in adult patients with hypokalaemia of renal origin suggesting Gitelman syndrome. Eur. J. Endocrinol. 2011, 165, 665–673.
- Calò, L.A.; Davis, P.A. Are the clinical presentations (Phenotypes) of gitelman’s and bartter’s syndromes gene mutations driven by their effects on intracellular ph, their “ph” enotype? Int. J. Mol. Sci. 2020, 21, 5660.
- Calò, L.A.; Davis, P.A.; Rossi, G.P. Understanding themechanisms of angiotensin II signaling involved in hypertension and its long-term sequelae: Insights from Bartter’s and Gitelman’s syndromes, humanmodels of endogenous angiotensin II signaling antagonism. J. Hypertens. 2014, 32, 2109–2119.
- Melander, O.; Orho-Melander, M.; Bengtsson, K.; Lindblad, U.; Råstam, L.; Groop, L.; Hulthén, U.L. Genetic variants of thiazide-sensitive NaCl-cotransporter in Gitelman’s syndrome and primary hypertension. Hypertension 2000, 36, 389–394.
- Berry, M.R.; Robinson, C.; Karet Frankl, F.E. Unexpected clinical sequelae of Gitelman syndrome: Hypertension in adulthood is common and females have higher potassium requirements. Nephrol. Dial. Transplant. 2013, 28, 1533–1542.
- Evans, R.D.R.; Antonelou, M.; Sathiananthamoorthy, S.; Rega, M.; Henderson, S.; Ceron-Gutierrez, L.; Barcenas-Morales, G.; Müller, C.A.; Doffinger, R.; Walsh, S.B.; et al. Inherited salt-losing tubulopathies are associated with immunodeficiency due to impaired IL-17 responses. Nat. Commun. 2020, 11, 1–19.
- Fujimura, J.; Nozu, K.; Yamamura, T.; Minamikawa, S.; Nakanishi, K.; Horinouchi, T.; Nagano, C.; Sakakibara, N.; Nakanishi, K.; Shima, Y.; et al. Clinical and Genetic Characteristics in Patients With Gitelman Syndrome. Kidney Int. Rep. 2019, 4, 119–125.
- Blanchard, A.; Vallet, M.; Dubourg, L.; Hureaux, M.; Allard, J.; Haymann, J.P.; de la Faille, R.; Arnoux, A.; Dinut, A.; Bergerot, D.; et al. Resistance to insulin in patients with gitelman syndrome and a subtle intermediate phenotype in heterozygous carriers: A cross-sectional study. J. Am. Soc. Nephrol. 2019, 30, 1534–1545.
- Ren, H.; Qin, L.; Wang, W.; Ma, J.; Zhang, W.; Shen, P.Y.; Shi, H.; Li, X.; Chen, N. Abnormal glucose metabolism and insulin sensitivity in Chinese patients with Gitelman syndrome. Am. J. Nephrol. 2013, 37, 152–157.
- Han, Y.; Zhao, X.; Wang, S.; Wang, C.; Tian, D.; Lang, Y.; Bottillo, I.; Wang, X.; Shao, L. Eleven novel SLC12A1 variants and an exonic mutation cause exon skipping in Bartter syndrome type I. Endocrine 2020, 64, 708–718.
- George, A.L.; Neilson, E.G. Biología celular y molecular de los riñones. In Harrison. Principios de Medicina Interna, 19th ed.; Kasper, D., Fauci, A., Hauser, S., Longo, D., Jameson, J.L., Loscalzo, J., Eds.; McGraw-Hill Education: New York, NY, USA, 2019.
- Brochard, K.; Boyer, O.; Blanchard, A.; Loirat, C.; Niaudet, P.; MacHer, M.A.; Deschenes, G.; Bensman, A.; Decramer, S.; Cochat, P.; et al. Phenotype-genotype correlation in antenatal and neonatal variants of Bartter syndrome. Nephrol. Dial. Transplant. 2009, 24, 1455–1464.
- Mourani, C.C.; Sanjad, S.A.; Akatcherian, C.Y. Bartter syndrome in a neonate: Early treatment with indomethacin. Pediatr. Nephrol. 2000, 14, 143–145.
- Finer, G.; Shalev, H.; Birk, O.S.; Galron, D.; Jeck, N.; Sinai-Treiman, L.; Landau, D. Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J. Pediatr. 2003, 142, 318–323.
- Gamba, G.; Friedman, P.A. Thick ascending limb: The Na+:K+:2Cl− co-transporter, NKCC2, and the calcium-sensing receptor, CaSR. Pflugers Arch. Eur. J. Physiol. 2009, 458, 61–76.
- Mutig, K.; Kahl, T.; Saritas, T.; Godes, M.; Persson, P.; Bates, J.; Raffi, H.; Rampoldi, L.; Uchida, S.; Hille, C.; et al. Activation of the bumetanide-sensitive Na +,K +, 2Cl -Cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J. Biol. Chem. 2011, 286, 30200–30210.
- Schiano, G.; Glaudemans, B.; Olinger, E.; Goelz, N.; Müller, M.; Loffing-Cueni, D.; Deschenes, G.; Loffing, J.; Devuyst, O. The Urinary Excretion of Uromodulin is Regulated by the Potassium Channel ROMK. Sci. Rep. 2019, 9, 1–12.
- Marcoux, A.A.; Tremblay, L.E.; Slimani, S.; Fiola, M.J.; Mac-Way, F.; Garneau, A.P.; Isenring, P. Molecular characteristics and physiological roles of Na+–K+–Cl− cotransporter 2. J. Cell. Physiol. 2021, 236, 1712–1729.
- Grill, A.; Schießl, I.M.; Gess, B.; Fremter, K.; Hammer, A.; Castrop, H. Salt-losing nephropathy in mice with a null mutation of the Clcnk2 gene. Acta Physiol. 2016, 218, 198–211.
- Hennings, J.C.; Andrini, O.; Picard, N.; Paulais, M.; Huebner, A.K.; Cayuqueo, I.K.L.; Bignon, Y.; Keck, M.; Cornière, N.; Böhm, D.; et al. The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J. Am. Soc. Nephrol. 2017, 28, 209–217.
- Pérez-Rius, C.; Castellanos, A.; Gaitán-Peñas, H.; Navarro, A.; Artuch, R.; Barrallo-Gimeno, A.; Estévez, R. Role of zebrafish ClC-K/barttin channels in apical kidney chloride reabsorption. J. Physiol. 2019, 597, 3969–3983.
- Nozu, K.; Inagaki, T.; Fu, X.J.; Nozu, Y.; Kaito, H.; Kanda, K.; Sekine, T.; Igarashi, T.; Nakanishi, K.; Yoshikawa, N.; et al. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J. Med. Genet. 2008, 45, 182–186.
- Estévez, R.; Boettger, T.; Stein, V.; Birkenhäger, R.; Otto, E.; Hildebrandt, F.; Jentsch, T.J. Barttin is a Cl− channel β-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 2001, 414, 558–561.
- Matsumura, Y.; Uchida, S.; Kondo, Y.; Miyazaki, H.; Ko, S.B.H.; Hayama, A.; Morimoto, T.; Liu, W.; Arisawa, M.; Sasaki, S.; et al. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat. Genet. 1999, 21, 95–98.
- Kruegel, J.; Rubel, D.; Gross, O. Alport syndrome—Insights from basic and clinical research. Nat. Rev. Nephrol. 2013, 9, 170–178.
- Rost, S.; Bach, E.; Neuner, C.; Nanda, I.; Dysek, S.; Bittner, R.E.; Keller, A.; Bartsch, O.; Mlynski, R.; Haaf, T.; et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur. J. Hum. Genet. 2014, 22, 208–215.
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076.
- Quigley, R.; Saland, J.M. Transient antenatal Bartter’s Syndrome and X-linked polyhydramnios: Insights from the genetics of a rare condition. Kidney Int. 2016, 90, 721–723.
- Allison, S.J. Renal physiology: MAGED2 mutations in transient antenatal Bartter syndrome. Nat. Rev. Nephrol. 2016, 12, 377.
- Bakhos-douaihy, D.; Seaayfan, E.; Demaretz, S.; Komhoff, M.; Laghmani, K. Differential effects of stch and stress—Inducible hsp70 on the stability and maturation of nkcc2. Int. J. Mol. Sci. 2021, 7, 2207.
- Donnelly, B.F.; Needham, P.G.; Snyder, A.C.; Roy, A.; Khadem, S.; Brodsky, J.L.; Subramanya, A.R. Hsp70 and Hsp90 multichaperone complexes sequentially regulate thiazide-sensitive cotransporter endoplasmic reticulum-associated degradation and biogenesis. J. Biol. Chem. 2013, 288, 13124–13135.
- Needham, P.G.; Mikoluk, K.; Dhakarwal, P.; Khadem, S.; Snyder, A.C.; Subramanya, A.R.; Brodsky, J.L. The thiazide-sensitive NaCl cotransporter is targeted for chaperone-dependent endoplasmic reticulum-associated degradation. J. Biol. Chem. 2011, 286, 43611–43621.
- Rosenbaek, L.L.; Rizzo, F.; Wu, Q.; Rojas-Vega, L.; Gamba, G.; MacAulay, N.; Staub, O.; Fenton, R.A. The thiazide sensitive sodium chloride co-transporter NCC is modulated by site-specific ubiquitylation. Sci. Rep. 2017, 7.
- Legrand, A.; Treard, C.; Roncelin, I.; Dreux, S.; Bertholet-Thomas, A.; Broux, F.; Bruno, D.; Decramer, S.; Deschenes, G.; Djeddi, D.; et al. Prevalence of novel MAGED2 mutations in antenatal bartter syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 242–250.
- Ellison, D.H.; Terker, A.S.; Gamba, G. Potassium and its discontents: New insight, new treatments. J. Am. Soc. Nephrol. 2016, 27, 981–989.
- Loffing, J.; Vallon, V.; Loffing-Cueni, D.; Aregger, F.; Richter, K.; Pietri, L.; Bloch-Faure, M.; Hoenderop, J.G.J.; Shull, G.E.; Meneton, P.; et al. Altered renal distal tubule structure and renal Na+ and Ca 2+ handling in a mouse model for Gitelman’s syndrome. J. Am. Soc. Nephrol. 2004, 15, 2276–2288.
- Wang, L.; Zhang, C.; Su, X.; Lin, D.H.; Wang, W. Caveolin-1 deficiency inhibits the basolateral K+ channels in the distal convoluted tubule and impairs renal K+ and Mg2+ transport. J. Am. Soc. Nephrol. 2015, 26, 2678–2690.
- Giebisch, G. Renal Potassium Channels: Function, Regulation, and Structure. In Proceedings of the Kidney International; Blackwell Publishing Inc.: Hoboken, NJ, USA, 2001; Volume 60, pp. 436–445.
- Waldegger, S.; Jentsch, T.J. Functional and structural analysis of ClC-K chloride channels involved in renal disease. J. Biol. Chem. 2000, 275, 24527–24533.
- Li, D.; Tian, L.; Hou, C.; Kim, C.E.; Hakonarson, H.; Levine, M.A. Association of mutations in SLC12A1 encoding the NKCC2 cotransporter with neonatal primary hyperparathyroidism. J. Clin. Endocrinol. Metab. 2016, 101, 2196–2200.
- Wongsaengsak, S.; Vidmar, A.P.; Addala, A.; Kamil, E.S.; Sequeira, P.; Fass, B.; Pitukcheewanont, P. A novel SLC12A1 gene mutation associated with hyperparathyroidism, hypercalcemia, nephrogenic diabetes insipidus, and nephrocalcinosis in four patients. Bone 2017, 97, 121–125.
- Chen, Q.; Wang, X.; Min, J.; Wang, L.; Mou, L. Kidney stones and moderate proteinuria as the rare manifestations of Gitelman syndrome. BMC Nephrol. 2021, 22, 12.
- Demoulin, N.; Aydin, S.; Cosyns, J.P.; Dahan, K.; Cornet, G.; Auberger, I.; Loffing, J.; Devuyst, O. Gitelman syndrome and glomerular proteinuria: A link between loss of sodium-chloride cotransporter and podocyte dysfunction? Nephrol. Dial. Transplant. 2014, 29, iv117–iv120.
- Walsh, P.R.; Tse, Y.; Ashton, E.; Iancu, D.; Jenkins, L.; Bienias, M.; Kleta, R.; Van’t Hoff, W.; Bockenhauer, D. Clinical and diagnostic features of Bartter and Gitelman syndromes. Clin. Kidney J. 2018, 11, 302–309.
- Bettinelli, A.; Borsa, N.; Bellantuono, R.; Syrèn, M.L.; Calabrese, R.; Edefonti, A.; Komninos, J.; Santostefano, M.; Beccaria, L.; Pela, I.; et al. Patients With Biallelic Mutations in the Chloride Channel Gene CLCNKB: Long-Term Management and Outcome. Am. J. Kidney Dis. 2007, 49, 91–98.
- Stokman, M.F.; Renkema, K.Y.; Giles, R.H.; Schaefer, F.; Knoers, N.V.A.M.; van Eerde, A.M. The expanding phenotypic spectra of kidney diseases: Insights from genetic studies. Nat. Rev. Nephrol. 2016, 12, 472–483.
- Konrad, M.; Nijenhuis, T.; Ariceta, G.; Bertholet-Thomas, A.; Calo, L.A.; Capasso, G.; Emma, F.; Schlingmann, K.P.; Singh, M.; Trepiccione, F.; et al. Diagnosis and management of Bartter syndrome: Executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int. 2021, 99, 324–335.
- Knoers, N.V.A.M. Gitelman syndrome. Adv. Chronic Kidney Dis. 2006, 13, 148–154.
- Ranade, V.V.; Somberg, J.C. Bioavailability and pharmacokinetics of magnesium after administration of magnesium salts to humans. Am. J. Ther. 2001, 8, 345–357.
- Jain, G.; Ong, S.; Warnock, D.G. Genetic disorders of potassium homeostasis. Semin. Nephrol. 2013, 33, 300–309.
- Beck Laurence, H.J.; Salant, D.J. Enfermedades tubulointersticiales del riñón. In Harrison. Principios de Medicina Interna, 20th ed.; Jameson, J.L., Fauci, A.S., Kasper, D.L., Hauser, S.L., Longo, D.L., Loscalzo, J., Eds.; McGraw-Hill Education: New York, NY, USA, 2018.
- Tarnawski, A.S.; Jones, M.K. Inhibition of angiogenesis by NSAIDs: Molecular mechanisms and clinical implications. J. Mol. Med. 2003, 81, 627–636.
- Suleyman, H.; Cadirci, E.; Albayrak, A.; Halici, Z. Nimesulide is a Selective COX-2 Inhibitory, Atypical Non-Steroidal Anti-Inflammatory Drug. Curr. Med. Chem. 2008, 15, 278–283.
- Fulchiero, R.; Seo-Mayer, P. Bartter Syndrome and Gitelman Syndrome. Pediatr. Clin. N. Am. 2019, 66, 121–134.
- Mackie, F.E.; Hodson, E.M.; Roy, L.P.; Knight, J.F. Neonatal Bartter syndrome—Use of indomethacin in the newborn period and prevention of growth failure. Pediatr. Nephrol. 1996, 10, 756–758.
- Gasongo, G.; Greenbaum, L.A.; Niel, O.; Kwon, T.; Macher, M.A.; Maisin, A.; Baudouin, V.; Dossier, C.; Deschênes, G.; Hogan, J. Effect of nonsteroidal anti-inflammatory drugs in children with Bartter syndrome. Pediatr. Nephrol. 2019, 34, 679–684.
- Larkins, N.; Wallis, M.; McGillivray, B.; Mammen, C. A severe phenotype of Gitelman syndrome with increased prostaglandin excretion and favorable response to indomethacin. Clin. Kidney J. 2014, 7, 306–310.
- Blanchard, A.; Vargas-Poussou, R.; Vallet, M.; Caumont-Prim, A.; Allard, J.; Desport, E.; Dubourg, L.; Monge, M.; Bergerot, D.; Baron, S.; et al. Indomethacin, amiloride, or eplerenone for treating hypokalemia in Gitelman syndrome. J. Am. Soc. Nephrol. 2015, 26, 468–475.
- Brater, D.C. Anti-inflammatory agents and renal function. Semin. Arthritis Rheum. 2002, 32, 33–42.
- Kleinknecht, D. Interstitial nephritis, the nephrotic syndrome, and chronic renal failure secondary to nonsteroidal anti-inflammatory drugs. Semin. Nephrol. 1995, 15, 228–235.
- Marlicz, W.; Łoniewski, I.; Grimes, D.S.; Quigley, E.M. Nonsteroidal anti-inflammatory drugs, proton pump inhibitors, and gastrointestinal injury: Contrasting interactions in the stomach and small Intestine. Mayo Clin. Proc. 2014, 89, 1699–1709.
- The Hospital for Sick Children: Indomethacin, Oral Suspension. Available online: https://www.sickkids.ca/en/care-services/for-health-care-providers/pharmacy/ (accessed on 18 October 2021).
- Dembo, G.; Park, S.B.; Kharasch, E.D. Central nervous system concentrations of cyclooxygenase-2 inhibitors in humans. Anesthesiology 2005, 102, 409–415.
- Solomon, D.H.; Schneeweiss, S.; Glynn, R.J.; Kiyota, Y.; Levin, R.; Mogun, H.; Avorn, J. Relationship between Selective Cyclooxygenase-2 Inhibitors and Acute Myocardial Infarction in Older Adults. Circulation 2004, 109, 2068–2073.
- Baigent, C.; Bhala, N.; Emberson, J.; Merhi, A.; Abramson, S.; Arber, N.; Baron, J.A.; Bombardier, C.; Cannon, C.; Farkouh, M.E.; et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: Meta-analyses of individual participant data from randomised trials. Lancet 2013, 382, 769–779.
- Jackson, E.K. Fármacos que afectan la función excretora renal. In Goodman & Gilman: Las Bases Farmacológicas, De La Terapéutic, 13th ed.; Brunton, L.L., Chabner, B.A., Knollmann, B.C., Eds.; McGraw-Hill Education: New York, NY, USA, 2019.
- Perazella, M.A. Trimethoprim is a potassium-sparing diuretic like amiloride and causes hyperkalemia in high-risk patients. Am. J. Ther. 1997, 4, 343–348.
- Plumb, L.A.; van’t Hoff, W.; Kleta, R.; Reid, C.; Ashton, E.; Samuels, M.; Bockenhauer, D. Renal apnoea: Extreme disturbance of homoeostasis in a child with Bartter syndrome type IV. Lancet 2016, 388, 631–632.
- Schepkens, H.; Lameire, N. Gitelman’s syndrome: An overlooked cause of chronic hypokalemia and hypomagnesemia in adults. Acta Clin. Belg. 2001, 56, 248–254.
- Koudsi, L.; Nikolova, S.; Mishra, V. Management of a severe case of Gitelman syndrome with poor response to standard treatment. BMJ Case Rep. 2016, 1–3.
- Griffing, G.T.; Komanicky, P.; Aurecchis, S.A.; Sindler, B.H.; Melby, J.C. Amiloride in Bartter’s syndrome. Clin. Pharmacol. Ther. 1982, 31, 713–718.
- Griffing, G.T.; Melby, J.C. The therapeutic use of a new potassium-sparing diuretic, amiloride, and a converting enzyme inhibitor, mk-421, in preventing hypokalemia associated with primary and secondary hyperaldosteronism. Clin. Exp. Hypertens. 1983, A5, 779–801.
- Griffing, G.T.; Aurecchia, S.A.; Sindler, B.H.; Melby, J.C. The Effect of Amiloride on the Renin—Aldosterone System in Primary Hyperaldosteronism and Bartter’s Syndrome. J. Clin. Pharmacol. 1982, 22, 505–512.
- Luqman, A.; Kazmi, A.; Wall, B.M. Bartter’s syndrome in pregnancy: Review of potassium homeostasis in gestation. Am. J. Med. Sci. 2009, 338, 500–504.
- Deruelle, P.; Dufour, P.; Magnenant, E.; Courouble, N.; Puech, F. Maternal Bartter’s syndrome in pregnancy treated by amiloride. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 115, 106–107.
- Federal Register: Content and Format of Labeling for Human Prescription Drug and Biological Products. Requirements for Pregnancy and Lactation Labeling. Available online: https://www.federalregister.gov/documents/2008/05/29/E8-11806/content-and-format-of-labeling-for-human-prescription-drug-and-biological-products-requirements-for (accessed on 24 May 2021).
- Santos, F.; Gil-Peña, H.; Blázquez, C.; Coto, E. Gitelman syndrome: A review of clinical features, genetic diagnosis and therapeutic management. Expert Opin. Orphan Drugs 2016, 4, 1005–1009.
- Morales, J.M.; Ruilope, L.M.; Praga, M.; Coto, A.; Alcazar, J.M.; Prieto, C.; Nieto, J.; Rodicio, J.L. Long-term enalapril therapy in Bartter’s syndrome. Nephron 1988, 48, 327.
- Hené, R.J.; Koomans, H.A.; Mees, E.J.D.; Stolpe, A.; Verhoef, G.E.G.; Boer, P. Correction of Hypokalemia in Bartter’s Syndrome by Enalapril. Am. J. Kidney Dis. 1987, 9, 200–205.
- Nascimento, C.L.P.; Garcia, C.L.; Schvartsman, B.G.S.; Vaisbich, M.H. Treatment of Bartter syndrome. Unsolved issue. J. Pediatr. 2014, 90, 512–517.
- Jest, P.; Pedersen, K.E.; Klitgaard, N.A.; Thomsen, N.; Kjaer, K.; Simonsen, E. Angiotensin-converting enzyme inhibition as a therapeutic principle in Bartter’s syndrome. Eur. J. Clin. Pharmacol. 1991, 41, 303–305.
- Álvarez-Nava, F.; Lanes, R. GH/IGF-1 signaling and current knowledge of epigenetics; A review and considerations on possible therapeutic options. Int. J. Mol. Sci. 2017, 18, 1624.
- Ficha Tecnica Genotonorm Kabipen 12 mg Polvo Y Disolvente Para Solucion Inyectable. Available online: https://cima.aemps.es/cima/dochtml/ft/60117/FT_60117.html (accessed on 4 May 2021).
- Cook, D.M.; Rose, S.R. A review of guidelines for use of growth hormone in pediatric and transition patients. Pituitary 2012, 15, 301–310.
- Drube, J.; Wan, M.; Bonthuis, M.; Wühl, E.; Bacchetta, J.; Santos, F.; Grenda, R.; Edefonti, A.; Harambat, J.; Shroff, R.; et al. Clinical practice recommendations for growth hormone treatment in children with chronic kidney disease. Nat. Rev. Nephrol. 2019, 15, 577–589.
- Collett-Solberg, P.F.; Ambler, G.; Backeljauw, P.F.; Bidlingmaier, M.; Biller, B.M.K.; Boguszewski, M.C.S.; Cheung, P.T.; Choong, C.S.Y.; Cohen, L.E.; Cohen, P.; et al. Diagnosis, Genetics, and Therapy of Short Stature in Children: A Growth Hormone Research Society International Perspective. Horm. Res. Paediatr. 2019, 92, 1–14.
- Yuen, K.C.J.; Biller, B.M.K.; Radovick, S.; Carmichael, J.D.; Jasim, S.; Pantalone, K.M.; Hoffman, A.R. American Association of Clinical endocrinologists and American College of Endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr. Pract. 2019, 25, 1191–1232.
- Touyz, R.M. Magnesium in clinical medicine. Front. Biosci. 2004, 9, 1278–1293.
- Sackett, D.L.; Haynes, R.B.; Tugwell, P. Clinical Epidemiology: A Basic Science for Clinical Medicine; Little, Brown & Co.: Boston, MA, USA, 1985; p. 370. ISBN 0-316-76595-3.
- Ariceta, G.; Rodríguez-Soriano, J. Inherited Renal Tubulopathies Associated With Metabolic Alkalosis: Effects on Blood Pressure. Semin. Nephrol. 2006, 26, 422–433.
- Shalev, H.; Ohali, M.; Kachko, L.; Landau, D. The neonatal variant of Bartter syndrome and deafness: Preservation of renal function. Pediatrics 2003, 112, 628–633.
- Jeck, N.; Reinalter, S.C.; Henne, T.; Marg, W.; Mallmann, R.; Pasel, K.; Vollmer, M.; Klaus, G.; Leonhardt, A.; Seyberth, H.W.; et al. Hypokalemic salt-losing tubulopathy with chronic renal failure and sensorineural deafness. Pediatrics 2001, 108, e5.
- ClinicalTrials.gov. Bartter Syndrome. Available online: https://clinicaltrials.gov/ct2/results?cond=bartter&term=&cntry=&state=&city=&dist= (accessed on 27 May 2021).
- ClinicalTrials.gov. Gitelman Syndrome. Available online: https://clinicaltrials.gov/ct2/results?cond=gitelman&draw=2&rank=3#rowId2 (accessed on 27 May 2021).
- Diamox® (Acetazolamide Extended-Release Capsules). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/12945s037,038lbl.pdf (accessed on 27 May 2021).
- Mazaheri, M.; Assadi, F.; Sadeghi-Bojd, S. Adjunctive acetazolamide therapy for the treatment of Bartter syndrome. Int. Urol. Nephrol. 2020, 52, 121–128.
- Blanchard, A.; Tabard, S.B.; Lamaziere, A.; Bergerot, D.; Zhygalina, V.; Lorthioir, A.; Jacques, A.; Hourton, D.; Azizi, M.; Crambert, G. Adrenal adaptation in potassium-depleted men: Role of progesterone? Nephrol. Dial. Transplant. 2020, 35, 1901–1908.

