Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Qianqian Ding | + 3081 word(s) | 3081 | 2021-09-23 05:10:55 | | | |
| 2 | Qianqian Ding | Meta information modification | 3081 | 2021-10-24 13:06:09 | | | | |
| 3 | Lindsay Dong | Meta information modification | 3081 | 2021-10-26 05:36:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ding, Q. Mitochondria in Maturation of Cardiomyocytes. Encyclopedia. Available online: https://encyclopedia.pub/entry/15318 (accessed on 28 June 2026).
Ding Q. Mitochondria in Maturation of Cardiomyocytes. Encyclopedia. Available at: https://encyclopedia.pub/entry/15318. Accessed June 28, 2026.
Ding, Qianqian. "Mitochondria in Maturation of Cardiomyocytes" Encyclopedia, https://encyclopedia.pub/entry/15318 (accessed June 28, 2026).
Ding, Q. (2021, October 24). Mitochondria in Maturation of Cardiomyocytes. In Encyclopedia. https://encyclopedia.pub/entry/15318
Ding, Qianqian. "Mitochondria in Maturation of Cardiomyocytes." Encyclopedia. Web. 24 October, 2021.
Copy Citation
Cardiomyocytes obtained from pluripotent stem cells (PSCs)differentiation culture are regarded as immature structurally, electrophysiologically, metabolically, and functionally. Mitochondria are organelles responsible for various cellular functions such as energy metabolism, different catabolic and anabolic processes, calcium fluxes, and various signaling pathways. Cells can respond to cellular needs to increase the mitochondrial mass by mitochondrial biogenesis. On the other hand, cells can also degrade mitochondria through mitophagy. Mitochondria are also dynamic organelles that undergo continuous fusion and fission events.
cardiomyocytes maturation
mitochondria
mitochondrial biogenesis
mitochondrial dynamics
mitophagy
1. Process of Cardiomyocyte Differentiation and Maturation
The mammalian heart is the first differentiated and functional organ in the developing embryo. The heart originates from the cells of the early embryonic mesoderm, which emerges from the primitive streak during gastrulation. The myocardial progenitor cells migrate from the primitive streak to the anterior of the embryo and form the cardiac crescent. The early cardiac tube forms through the fusion of the cardiac crescent at the midline. The looping of the cardiac tube and expansion of the myocardium further leads to the formation of recognizable cardiac chambers [1][2]. The mammalian heart is composed of several major cell types: cardiomyocytes (CMs), smooth muscle cells, endothelial cells, as well as fibroblasts [3]. During cardiac development, there are two sources of cardiac progenitor cells (CPCs), namely the first heart field (FHF) and the second heart field (SHF) [4][5]. The CPCs of the FHF give rise to the left ventricle, whereas the CPCs of the SHF develop into a large part of the definitive heart including atria and inflow tract at the venous pole and right ventricle and outflow tract at the arterial pole [1][6][7].
The heart undergoes a complex differentiation and maturation process throughout embryonic and postnatal stages. The growth of the embryonic heart is mainly due to CM proliferation, while postnatal CMs lose the ability to proliferate soon after birth, and the continued increase in the heart mass is driven by the enlargement of existing CMs [8][9][10]. The differentiation and morphogenesis of mammalian hearts have been focal points for developmental cardiology. However, the maturation of the heart has been less studied until recently. Maturation is the last phase of heart development, a process in which immature CMs transit to mature CMs in terms of cell structure, gene expression, electrophysiological properties, and energy metabolism.
The reason for the increased attention towards the maturation of CMs is the emerging pluripotent stem cell (PSC)-based regenerative therapy. Since the loss of CMs after heart injury is irreversible, PSC-derived CMs (PSC-CMs) have shown great promise in heart repair and functional improvement [11]. Although methods to differentiate PSCs into CMs have been well-established and those PSC-CMs show similar molecular, electrical, mechanical, and ultrastructural features to CMs, their fetal-like phenotypes limit the successful application of these cells for research and medicine [12][13]. Indeed, the following changes occur as CMs mature ex vivo: (i) adult cardiac genes instead of fetal cardiac genes are expressed; (ii) cell size increases and cell shape changes from circular to rod shape; (iii) sarcomere length increases and t-tubules are formed; (iv) electrophysiologically, action potential (AP) upstroke becomes faster, AP duration increases, maximum diastolic potential becomes more hyperpolarized, and diastolic depolarization slope decreases until the cells become electrically quiescent yet excitable; (v) mitochondria shape changes from small and round to firstly slender and long and subsequently ovular; (vi) metabolically, cells rely on oxidative phosphorylation (OXPHOS) instead of glycolysis for ATP production; and (vii) metabolic substrate changes from glucose to fatty acid [14][15] (Figure 1). However, PSC-CMs cultured under the conventional protocol are known to be largely immature with regard to the above features [14][15]. Although previous studies have tried to enhance PSC-CM maturation using different strategies, PSC-CMs are still regarded as unlike adult CMs. Specifically for mitochondria, mitochondria in PSC-CMs occupy a smaller cellular volume than that in adult CMs. Moreover, PSC-CMs rely on glycolysis rather than fatty acid β-oxidation (FAO) for ATP generation [16][17]. Thus, the challenge in front of us is to develop strategies to reach a higher degree of PSC-CM maturation in vitro.
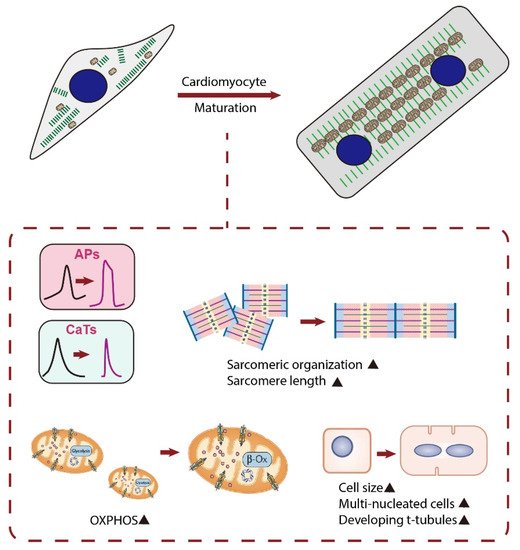
Figure 1. Schematic diagram showing major changes in characteristics of CMs during maturation. Matured CMs have an increased cell size, and the cell shape changes from circular to rod-shaped. Matured CMs have an improved alignment of sarcomeres, longer sarcomeres, and existence of t-tubules and become multi-nucleated. They also have enhanced calcium handling, electrophysiology, and metabolism. APs, action potentials; CaTs, calcium transients; OXPHOS, oxidative phosphorylation; β-Ox, β-oxidation.
2. Mitochondrial Biogenesis and Cardiac Maturation
As we mentioned before, generating more physiologically mature CMs is vital for drug screening, disease modeling, and therapeutic purposes. As reported by many studies, structural and functional maturation of CMs is always accompanied by more mature energy metabolism. During early cardiac development, glycolysis is a major source of energy for CM proliferation. Perinatal mitochondrial biogenic surge in CMs is accompanied by a metabolic shift from using glucose to fatty acids for ATP generation [18]. As CMs mature and become terminally differentiated, mitochondrial oxidative capacity increases, with FAO becoming the major source of energy [19]. This shift is important to fulfill the increased cardiac workload as FAO is much more efficient than glycolysis for ATP generation. As development proceeds, mitochondria occupy ~20–40% of adult CM volume [20]. It has been documented that in PSC-CMs, the changes in energy metabolism have important impacts on the ability of CMs to proliferate during early cardiac development, as well as when CMs terminally differentiate during later development [21]. A previous study reported that an appropriate metabolic shift from aerobic glycolysis to OXPHOS would in turn improve metabolic and functional maturation of human PSC-CMs (hPSC-CMs) [22]. Fatty acid supplementation boosted hPSC-CM maturation with enhanced calcium transient peak height and kinetics and increased AP upstroke velocity and membrane capacitance [23]. Similarly, maturation media designed to provide oxidative substrates adapted to the metabolic needs of human induced pluripotent stem cell (hiPSC)-derived CMs (hiPSC-CMs) improved the physiological function of hiPSC-CMs [24].
As the key regulator of mitochondrial biogenesis and metabolism, PGC-1α is supposed to help promote cardiac maturation. PGC-1α knockout (KO) mice were viable, but postnatal growth of the heart and slow-twitch skeletal muscle, organs with high mitochondrial energy demands, is blunted [25]. PGC-1β KO mice exhibit phenotypes that are very similar to PGC-1α KO mice [26]. However, PGC-1α/β double-knockout (DKO) mice died shortly after birth with small hearts, bradycardia, intermittent heart block, and a markedly reduced cardiac output, suggesting the possibility that PGC-1α and PGC-1β control a subset of overlapping targets and are, therefore, capable of compensating for the loss of the other factor [27]. Cardiac-specific ablation of PGC-1α and PGC-1β caused a set of maturational defects including reduced growth, a late fetal arrest in mitochondrial biogenesis, and persistence of a fetal pattern of gene expression, indicating that PGC-1 is indispensable for perinatal maturation of the heart [27]. Recently, it has been demonstrated that PGC-1α can promote the maturation of CMs derived from hESCs [28]. The activator of PGC-1α, ZLN005, upregulated the expressions of PGC-1α and mitochondrial function-related genes in hESC-CMs and induced more mature energy metabolism compared with the control group. In addition, ZLN005 treatment increased cell sarcomere length, improved cell calcium handling, and enhanced intercellular connectivity [28]. Single-cell gene network analysis showed that PGC-1 drives CM maturation via YAP1 and SF3B2 [29]. All these data indicate that PGC-1 plays as a multifaceted regulator coordinating cellular hypertrophy, contractility, and metabolism of CMs from immature to mature.
PGC-1α and PGC-1β serve to coactivate downstream transcriptional events by interacting with specific transcription factors, including the ERRs. Conditional gene disruption strategies were used by Sakamoto et al. to detect the role of ERRs in cardiac differentiation and maturation. They demonstrated that ERRα and ERRγ are necessary for normal postnatal cardiac developmental maturation. ERRγ functions as a direct transcriptional activator of metabolic and structural cardiac genes. Moreover, ERRα/γ suppress a subset of fetal and non-cardiac myocyte genes, including the fibroblast lineage [30].
Apart from the predominant role of nuclear genes, mitochondrial genetic systems are also required for mitochondrial biogenesis. Among them, TFAM and TFB2M are reported to be required for mitochondrial genome replication and transcription [31]. A recent study showed that TFAM inactivation by the CRE-Lox system controlled under cardiac-specific Nkx2.5 locus caused mitochondrial dysfunction and embryonic lethal myocardial hypoplasia. Neonatal TFAM inactivation by AAV9-cTnT-Cre caused progressive, lethal dilated cardiomyopathy, while postnatal TFAM inactivation and disruption of mitochondrial function did not impair CM maturation [32]. The failure to observe CM maturation defects in postnatal TFAM inactivation is possible if TFAM functions only at an earlier stage before birth. However, more detailed investigation is needed to elucidate the exact role of TFAM and TFB2M in CM maturation.
Taken together, these results showed that mitochondrial biogenesis is essential for CM maturation (Figure 2) and unveiled a new strategy to improve the maturation of PSC-CMs and therefore to generate more physiologically mature CMs for drug screening, disease modeling, and therapeutic purposes.
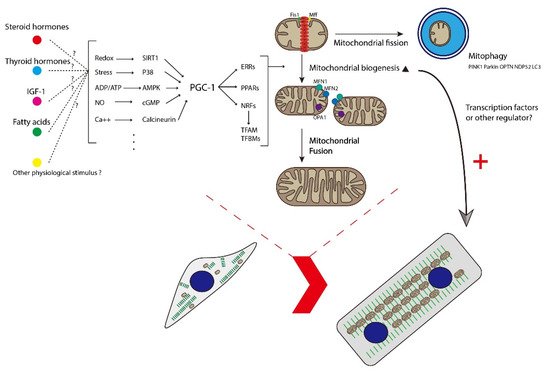
Figure 2. Schematic diagram showing how mitochondrial biogenesis, dynamics, and mitophagy participate in the maturation of CMs. Multiple stimuli activate PGC-1, leading to the coactivation of key transcription factors involved in several aspects of mitochondrial and cellular function. Mitochondria dynamically change their morphology through the cycle of fusion and fission. The main fusion factors are OPA1, MFN1, and MFN2, which bind to the IMM and OMM of mitochondria. Drp1 is a major fission factor that binds to OMM and forms a ring-like structure around mitochondria, leading to the separation of mitochondria into two. Mff and Fis1 function as adaptors to recruit Drp1 to the OMM. Mitochondrial fission and mitophagy function as quality control to segregate and degrade immature or damaged mitochondria and to provide materials for mitochondrial biogenesis. Healthy mitochondria tend to fuse together and are believed to function well. The maturation of healthy mitochondria further promotes the maturation of CMs at structural and functional levels. IGF-1, insulin-like growth factor 1; PGC-1, peroxisome proliferator-activated receptor γ coactivator 1; ERR, estrogen-related receptor; PPARs, peroxisome proliferator-activated receptors; NRF, nuclear respiratory factor; TFAM, transcription factor A, mitochondrial; TFBMs, mitochondrial transcription factor B; Fis1, fission protein-1; Mff, mitochondrial fission factor; OPA1, optic atrophy 1; MFN1, mitofusin 1; MFN2, mitofusin 2; PINK1, PTEN-induced kinase 1; OPTN, optineurin; NDP52, calcium binding and coiled-coil domain 2; LC3, microtubule-associated protein 1A/1B-light chain 3.
3. Mitophagy and Cardiac Maturation
After birth, the immature heart of a mammal is exposed to a different environment with relatively higher oxygen concentration and lower carbohydrate supplement [33][34]. Therefore, the metabolic transitions through which CMs switch to rely on FAO as primary energy source are needed [35]. This is not just an adaptative response; the growing organism requires a powerful, energy consuming heart with efficient ATP synthesis [36]. Immature fetal mitochondria showing few and sparse cristae for ETC are incapable of efficient ATP production; therefore, substitution or manipulation of mitochondria is needed. Although it has been conventionally believed that changes in mitochondrial gene expression at transcriptional level cause this transition [37], Gong et al. suggested that mitophagy-mediated replacement of fetal mitochondria is essential for this transition [38]. They interrupted Parkin-dependent mitophagy by generating transgenic mice with a mutant version of Mfn2 that can be temporally and spatially expressed in CMs starting from birth [38]. The mutant Mfn2 could inhibit mitochondrial translocation of Parkin but could still promote mitochondrial fusion as wild-type Mfn2 does [38]. Compared with mice expressing wild-type Mfn2, mutant Mfn2 mice persisted fetal mitochondrial morphology and immature mitochondrial function [38]. Perinatal mutant Mfn2 expression also failed the metabolic gene reprogramming and led to cardiomyopathy [38]. This study demonstrated the role of mitophagy during postnatal maturation of CMs. Since reliance on glycolysis as the primary energy source is one of the features of PSC-CMs, it is reasonable to investigate the role of mitophagy in the maturation of PSC-CMs [14]. Zhao et al. reported the investigation on mitophagy during maturation of ESC-CMs. They found that the promotional effect of glucocorticoids on the maturation of ESC-CMs may be related to Parkin-dependent mitophagy [39]. Interruption of mitophagy through both autophagy inhibition and knocking down of Parkin expression could block the promotional effect of glucocorticoid on ESC-CM maturation [39]. PINK1-Parkin-dependent mitophagy targeting depolarized mitochondria is thought to carry out quality control of mitochondria. The abovementioned evidence suggests that this pathway is not only a passive response after the appearance of dysfunctional mitochondria but also an available tool for programmed mitochondrial clearance. Thus, it might be reasonable to ask whether manipulating mitophagy could promote the maturation of CMs (Figure 2).
4. Mitochondrial Dynamics and Cardiac Maturation
Mitochondrial fission and fusion events transmit signaling messengers into changes of mitochondrial function, such as calcium buffering and metabolism within the cell. Mitochondrial dynamics have been implicated in a variety of biological processes including embryonic development. It has been proven by several groups that the deletion of proteins regulating mitochondrial dynamics, such as Mfn1, Mfn2, and Drp1, causes defects in cardiac development [40][41][42][43], suggesting that mitochondrial dynamics play vital roles in cardiogenesis.
To reveal the function of mitochondrial dynamics in cardiac development and maturation, researchers simultaneously ablated Mfn1 and Mfn2 in the embryonic or adult mice heart [40][44][45]. The study of Chen et al. showed that embryonic combined Mfn1/Mfn2 ablation was lethal after E9.5. Conditional combined Mfn1/Mfn2 ablation in adult hearts induced mitochondrial fragmentation, respiratory dysfunction, and rapidly progressive and lethal dilated cardiomyopathy [44]. Kasahara et al. also found that embryo lethality occurred after E9.5 in Mfn1/Mfn2 DKO mice; the DKO hearts were markedly hypoplastic, with biventricular wall thinning and poor trabeculation [40]. In addition, gene-trapping of Mfn2 or Opa1 in mouse ESCs impaired the differentiation of ESCs into CMs. The following mechanistic study showed that increased Ca2+-dependent calcineurin activity and Notch1 signaling might be responsible for the impaired ESC differentiation because fragmented mitochondria have decreased Ca2+ buffer ability, leading to increased cytosolic Ca2+ [40]. DKO mice with Mfn1 and Mfn2 being genetically inactivated in mid-gestation using a loxP/Myh6-cre approach were reported to have normal cardiac morphology and function at birth [45]. The mitochondria increased in number and appeared to be spherical and heterogeneous but exhibited normal electron density. By postnatal day 7, the mitochondrial number continued to increase, and many lost matrix components and membrane organization. The DKO mice developed dilated cardiomyopathy, and all died before postnatal day 16. Gene expression analysis showed that mitochondria biogenesis genes and mitophagy markers are altered, resulting in decreased mitochondrial biogenesis and hampered mitochondrial elimination [45]. All these studies indicate that mitochondrial fusion is essential for embryonic cardiac differentiation and postnatal cardiac development. In vitro study of hiPSC differentiation into CMs showed that enhanced mitochondrial fusion in hiPSCs significantly increased the expression of cardiac-specific genes, making mitochondrial fusion promoters promising molecular targets for generating lineages of the heart from hiPSCs for patient-specific regenerative medicine [46].
As the key factor of mitochondrial fission, the role of Drp1 in CM development was also determined. Drp1 was deleted by using a Myh6-Cre transgenic line [47]. Echocardiography at postnatal day 7 revealed that left ventricular function was significantly compromised, with decreased contraction and heart rate. All Drp1 KO mice died between postnatal day 9 and day 11. In addition, mitochondria interconnectivity increased, and respiration decreased in Drp1 KO CMs [48]. Similarly, muscle-specific Drp1 KO mice showed neonatal lethality due to dilated cardiomyopathy. The Drp1 ablation in heart and primary cultured CMs resulted in severe mtDNA nucleoid clustering and led to compromised mitochondrial respiration, which further led to immature myofibril assembly and defective CM hypertrophy [49]. On the contrary, the study of Hoque et al. demonstrated that changes in mitochondrial morphology from a small granular fragmented phenotype in PSCs to a filamentous reticular elongated network in differentiated CMs were detected during cardiac mesodermal differentiation and maturation. Interestingly, treatment of iPSCs with Mdivi-1, a pharmacological inhibitor of mitochondrial fission protein Drp1, during cardiac differentiation increased the percentage of beating embryoid bodies and expression of cardiac-specific genes. In addition, Drp1 gene silencing was accompanied by increased mitochondrial respiration and decreased aerobic glycolysis [50]. These results indicated that Drp1-mediated mitochondrial fission is indispensable for cardiac development, but a moderate shifting of mitochondrial morphology toward fusion by inhibition of Drp1 may help to promote cardiac differentiation and maturation with a metabolic shift from glycolysis towards OXPHOS, as fused and interconnected mitochondria are regarded to function well and exhibit enhanced metabolic capacity. Taken together, mitochondrial dynamics play important roles in cardiac development. Manipulating mitochondrial fission and fusion may be one of the possible ways to improve the maturation of CMs (Figure 2).
5. Future Direction
Interestingly, mitochondrial metabolites from the TCA cycle (e.g., acetyl-Co A, citrate, aconitate, α-ketoglutarate, succinate, fumarate, malate, oxaloacetate) are reported to control unique cellular function and cell fate [51][52]. The fact that the metabolic process is regulated by mitochondrial metabolites, such as NADH/NAD+, ATP/ADP, ATP/AMP, and succinate/α-ketoglutarate, indicated a non-negligible role of mitochondrial metabolites [51][53][54][55]. For instance, recent studies showed that mitochondrial metabolites can regulate cell function through modulating gene transcription and translation in cancer cells and immune effector cells [56][57][58][59]. In addition, several TCA metabolites, such as acetyl-CoA and succinate, have been demonstrated to participate in cardiovascular diseases including diabetes and ischemia-reperfusion injury [60][61][62]. For their potential role in cell fate determination, it was reported that in naïve embryonic stem cells, glutamine-derived α-ketoglutarate helps maintain a high α-ketoglutarate-to-succinate ratio, which is important for promoting histone/DNA demethylation and for maintaining pluripotency [63]. In addition, α-ketoglutarate was found to promote early differentiation of PSCs [64][65].
In addition, the physiological stimuli that could promote CM maturation through regulating mitochondrial biogenesis are in need of further exploration. The transition of metabolic reliance towards FAO after birth and the surge of various hormones in or after late gestation should be noticed. For instance, observation of dramatic rise in glucocorticoid levels shortly before birth led to the discovery of PGC-1α’s role as a downstream target of the glucocorticoid receptor in glucocorticoid-promoted maturation of the fetal heart [66]. Further investigation along this direction is needed.
Moreover, the interaction of cardiac transcription factors with PGC-1 may be tested. Similarly, increasing in mitochondrial amount and mass might generate signals which regulate maturation of CMs. Some signals including AMP-to-ATP ratio and NAD+-to-NADH ratio change simultaneously with the change in mitochondrial functions. Signaling pathways including AMPK, STAT3, and SIRT3 have been shown to be related to the retrograde signaling from mitochondria to nuclei. The positive involvement of cardiac transcription factors in these retrograde signaling pathways requires more detailed studies.
The role of Parkin-mediated mitophagy in regulating metabolic maturation during postnatal development of mouse heart has been revealed. However, this knowledge has not been sufficiently discussed or applied in the issue of PSC-CM immaturity. More attention should be paid to identifying different pathways of mitophagy in CMs and their potential contribution to promote maturation of PSC-CMs. In addition, a deeper investigation of upstream signaling, which may regulate mitophagy, may lead to a method to enhance the maturation of PSC-CMs.
References
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835.
- Fishman, M.C.; Chien, K.R. Fashioning the vertebrate heart: Earliest embryonic decisions. Development 1997, 124, 2099–2117.
- Garry, D.J.; Olson, E.N. A common progenitor at the heart of development. Cell 2006, 127, 1101–1104.
- Meilhac, S.M.; Esner, M.; Kelly, R.G.; Nicolas, J.F.; Buckingham, M.E. The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev. Cell 2004, 6, 685–698.
- Lescroart, F.; Chabab, S.; Lin, X.; Rulands, S.; Paulissen, C.; Rodolosse, A.; Auer, H.; Achouri, Y.; Dubois, C.; Bondue, A.; et al. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat. Cell Biol. 2014, 16, 829–840.
- Kelly, R.G. The second heart field. Curr. Top. Dev. Biol. 2012, 100, 33–65.
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart fields and cardiac morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a015750.
- Bugaisky, L.; Zak, R. Cellular growth of cardiac muscle after birth. Tex Rep. Biol Med. 1979, 39, 123–138.
- Oparil, S.; Bishop, S.P.; Clubb, F.J., Jr. Myocardial cell hypertrophy or hyperplasia. Hypertension 1984, 6 Pt 2, Iii38–43.
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell Cardiol. 1996, 28, 1737–1746.
- Yanamandala, M.; Zhu, W.; Garry, D.J.; Kamp, T.J.; Hare, J.M.; Jun, H.W.; Yoon, Y.S.; Bursac, N.; Prabhu, S.D.; Dorn, G.W., 2nd; et al. Overcoming the Roadblocks to Cardiac Cell Therapy Using Tissue Engineering. J. Am. Coll Cardiol. 2017, 70, 766–775.
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106.
- Maroli, G.; Braun, T. The long and winding road of cardiomyocyte maturation. Cardiovasc. Res. 2020, 117, 712–726.
- Yang, X.; Pabon, L.; Murry, C.E. Engineering adolescence: Maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2014, 114, 511–523.
- Jiang, Y.; Park, P.; Hong, S.M.; Ban, K. Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells: Current Strategies and Limitations. Mol. Cells 2018, 41, 613–621.
- Dai, D.F.; Danoviz, M.E.; Wiczer, B.; Laflamme, M.A.; Tian, R. Mitochondrial Maturation in Human Pluripotent Stem Cell Derived Cardiomyocytes. Stem. Cells Int. 2017, 2017, 5153625.
- Garbern, J.C.; Lee, R.T. Mitochondria and metabolic transitions in cardiomyocytes: Lessons from development for stem cell-derived cardiomyocytes. Stem Cell Res. 2021, 12, 177.
- Zhao, Q.; Sun, Q.; Zhou, L.; Liu, K.; Jiao, K. Complex Regulation of Mitochondrial Function During Cardiac Development. J. Am. Heart Assoc. 2019, 8, e012731.
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140.
- Schaper, J.; Meiser, E.; Stämmler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391.
- Morita, Y.; Tohyama, S. Metabolic Regulation of Cardiac Differentiation and Maturation in Pluripotent Stem Cells: A Lesson from Heart Development. Jma J. 2020, 3, 193–200.
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; van der Meer, P.; Serra, M.; et al. Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes by Inhibition of HIF1alpha and LDHA. Circ. Res. 2018, 123, 1066–1079.
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C., Jr.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem. Cell Rep. 2019, 13, 657–668.
- Feyen, D.A.M.; McKeithan, W.L.; Bruyneel, A.A.N.; Spiering, S.; Hormann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925.
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005, 3, e101.
- Lelliott, C.J.; Medina-Gomez, G.; Petrovic, N.; Kis, A.; Feldmann, H.M.; Bjursell, M.; Parker, N.; Curtis, K.; Campbell, M.; Hu, P.; et al. Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 2006, 4, e369.
- Lai, L.; Leone, T.C.; Zechner, C.; Schaeffer, P.J.; Kelly, S.M.; Flanagan, D.P.; Medeiros, D.M.; Kovacs, A.; Kelly, D.P. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008, 22, 1948–1961.
- Liu, Y.; Bai, H.; Guo, F.; Thai, P.N.; Luo, X.; Zhang, P.; Yang, C.; Feng, X.; Zhu, D.; Guo, J.; et al. PGC-1α activator ZLN005 promotes maturation of cardiomyocytes derived from human embryonic stem cells. Aging 2020, 12, 7411–7430.
- Murphy, S.A.; Miyamoto, M.; Kervadec, A.; Kannan, S.; Tampakakis, E.; Kambhampati, S.; Lin, B.L.; Paek, S.; Andersen, P.; Lee, D.I.; et al. PGC1/PPAR drive cardiomyocyte maturation at single cell level via YAP1 and SF3B2. Nat. Commun. 2021, 12, 1648.
- Sakamoto, T.; Matsuura, T.R.; Wan, S.; Ryba, D.M.; Kim, J.U.; Won, K.J.; Lai, L.; Petucci, C.; Petrenko, N.; Musunuru, K.; et al. A Critical Role for Estrogen-Related Receptor Signaling in Cardiac Maturation. Circ. Res. 2020, 126, 1685–1702.
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J. Biol. Chem. 2010, 285, 18129–18133.
- Zhang, D.; Li, Y.; Heims-Waldron, D.; Bezzerides, V.; Guatimosim, S.; Guo, Y.; Gu, F.; Zhou, P.; Lin, Z.; Ma, Q.; et al. Mitochondrial Cardiomyopathy Caused by Elevated Reactive Oxygen Species and Impaired Cardiomyocyte Proliferation. Circ. Res. 2018, 122, 74–87.
- Porter, G.A., Jr.; Hom, J.; Hoffman, D.; Quintanilla, R.; de Mesy Bentley, K.; Sheu, S.-S. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatr. Cardiol. 2011, 31, 75–81.
- Bartelds, B.; Knoester, H.; Smid, G.B.; Takens, J.; Visser, G.H.; Penninga, L.; Leij, F.R.v.d.; Beaufort-Krol, G.C.M.; Zijlstra, W.G.; Heymans, H.S.A.; et al. Perinatal Changes in Myocardial Metabolism in Lambs. Circulation 2000, 102, 926–931.
- Talman, V.; Teppo, J.; Pöhö, P.; Movahedi, P.; Vaikkinen, A.; Karhu, S.T.; Trošt, K.; Suvitaival, T.; Heikkonen, J.; Pahikkala, T.; et al. Molecular Atlas of Postnatal Mouse Heart Development. J. Am. Heart. Asso. 2018, 7, e010378.
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151.
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the fetal gene program. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198.
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, 6265.
- Zhou, R.; Li, J.; Zhang, L.; Cheng, Y.; Yan, J.; Sun, Y.; Wang, J.; Jiang, H. Role of Parkin-mediated mitophagy in glucocorticoid-induced cardiomyocyte maturation. Life Sci. 2020, 255, 117817.
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W., 2nd; Scorrano, L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 2013, 342, 734–737.
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol 2009, 11, 958–966.
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–286.
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991.
- Chen, Y.; Liu, Y.; Dorn, G.W., 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331.
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 2012, 111, 1012–1026.
- Lees, J.G.; Kong, A.M.; Chen, Y.C.; Sivakumaran, P.; Hernández, D.; Pébay, A.; Harvey, A.J.; Gardner, D.K.; Lim, S.Y. Mitochondrial Fusion by M1 Promotes Embryoid Body Cardiac Differentiation of Human Pluripotent Stem Cells. Stem. Cells Int. 2019, 2019, 6380135.
- Gaussin, V.; Van de Putte, T.; Mishina, Y.; Hanks, M.C.; Zwijsen, A.; Huylebroeck, D.; Behringer, R.R.; Schneider, M.D. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc. Natl. Acad. Sci. USA 2002, 99, 2878–2883.
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Höke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014, 33, 2798–2813.
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol. Cell Biol. 2015, 35, 211–223.
- Hoque, A.; Sivakumaran, P.; Bond, S.T.; Ling, N.X.Y.; Kong, A.M.; Scott, J.W.; Bandara, N.; Hernández, D.; Liu, G.S.; Wong, R.C.B.; et al. Mitochondrial fission protein Drp1 inhibition promotes cardiac mesodermal differentiation of human pluripotent stem cells. Cell Death Discov. 2018, 4, 39.
- Frezza, C. Mitochondrial metabolites: Undercover signalling molecules. Interface Focus 2017, 7, 20160100.
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Jones, A.W.; Yao, Z.; Vicencio, J.M.; Karkucinska-Wieckowska, A.; Szabadkai, G. PGC-1 family coactivators and cell fate: Roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion 2012, 12, 86–99.
- Fulco, M.; Sartorelli, V. Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle 2008, 7, 3669–3679.
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060.
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193.
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662.
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014, 7, 1679–1690.
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martínez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499.
- Pell, V.R.; Chouchani, E.T.; Frezza, C.; Murphy, M.P.; Krieg, T. Succinate metabolism: A new therapeutic target for myocardial reperfusion injury. Cardiovasc. Res. 2016, 111, 134–141.
- Ceperuelo-Mallafré, V.; Llauradó, G.; Keiran, N.; Benaiges, E.; Astiarraga, B.; Martínez, L.; Pellitero, S.; González-Clemente, J.M.; Rodríguez, A.; Fernández-Real, J.M.; et al. Preoperative Circulating Succinate Levels as a Biomarker for Diabetes Remission After Bariatric Surgery. Diabetes Care 2019, 42, 1956–1965.
- Kolwicz, S.C., Jr.; Olson, D.P.; Marney, L.C.; Garcia-Menendez, L.; Synovec, R.E.; Tian, R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 2012, 111, 728–738.
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416.
- Hwang, I.Y.; Kwak, S.; Lee, S.; Kim, H.; Lee, S.E.; Kim, J.H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H.; Jin, X.; et al. Psat1-Dependent Fluctuations in alpha-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metab. 2016, 24, 494–501.
- TeSlaa, T.; Chaikovsky, A.C.; Lipchina, I.; Escobar, S.L.; Hochedlinger, K.; Huang, J.; Graeber, T.G.; Braas, D.; Teitell, M.A. alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016, 24, 485–493.
- Rog-Zielinska, E.A.; Craig, M.A.; Manning, J.R.; Richardson, R.V.; Gowans, G.J.; Dunbar, D.R.; Gharbi, K.; Kenyon, C.J.; Holmes, M.C.; Hardie, D.G.; et al. Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: A role for PGC-1α. Cell Death Differ. 2015, 22, 1106–1116.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
3 times
(View History)
Update Date:
26 Oct 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No