Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manel Juan | + 2322 word(s) | 2322 | 2021-09-23 11:05:57 | | | |
| 2 | Lindsay Dong | Meta information modification | 2322 | 2021-10-26 11:39:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Juan, M. CAR-T Treatments in B-Cell Malignancies. Encyclopedia. Available online: https://encyclopedia.pub/entry/15240 (accessed on 27 June 2026).
Juan M. CAR-T Treatments in B-Cell Malignancies. Encyclopedia. Available at: https://encyclopedia.pub/entry/15240. Accessed June 27, 2026.
Juan, Manel. "CAR-T Treatments in B-Cell Malignancies" Encyclopedia, https://encyclopedia.pub/entry/15240 (accessed June 27, 2026).
Juan, M. (2021, October 21). CAR-T Treatments in B-Cell Malignancies. In Encyclopedia. https://encyclopedia.pub/entry/15240
Juan, Manel. "CAR-T Treatments in B-Cell Malignancies." Encyclopedia. Web. 21 October, 2021.
Copy Citation
B-cell malignancies, like leukemias and lymphomas, are neoplasms that emerge from the malignant proliferation of B cells. A cell-based immunotherapy, the chimeric antigen receptor (CAR) T-cells, has proven to improve the clinical outcome of relapsed/refractory HSCT B-cell lymphoproliferative disorders patients in clinical trials.
B-cell malignancies
CAR-T
1. B-Cell Lymphoproliferative Disorders and 1st Line of Treatment
B-cell malignancies are a diverse group of neoplasms that emerge from the malignant proliferation of B cells during their different stages of development [1], and they include lymphomas and leukemias [2]. The first line of treatment includes chemotherapy, but proposals of chemotherapy vary depending on the subtype of disorder and multiple patient factors [3], new chemoimmunotherapy drugs, such as ibrutinib and imatinib, as well as hematopoietic stem cell transplant (HSCT) [4][5][6][7].
2. CAR-Redirected T-Cells (CAR-T) Offer a New and Promising Cell-Based Immunotherapy
Immunotherapy, the most recent and effective cancer therapy, relies on using our own immune system to recognize and eliminate cancer cells. Beyond SCT, there are several types of additional immunotherapies: monoclonal antibodies, cancer vaccines, or the most successful ones so far, immune checkpoint inhibitors [8] and T-cell transfer therapy [9], including adoptive cell therapy using tumor-infiltrating lymphocytes (TILs) [10] or chimeric antigen receptor (CAR) T-cells [11].
Adoptive cell therapy depends on the capacity of the T-cell to fight and destroy cancer cells. Using gene transfer technologies (transfection of a gene inside cells through vectors or gene editing), it has been possible to genetically modify T-cells to stably express defective or “new” molecules. Although replacement of the wild-type gene in mutated T-cell precursors was the initial gene transfer proposal to treat some severe immunodeficiencies [12], the most revolutionary changes that gene therapy is providing on immune cells arrive from the modification of the T-cell surface by expression of antibody-derived chimeric molecules with binding domains that confer novel antigen recognition in a major histocompatibility complex (MHC)-independent manner. CARs are the most frequent application of these approaches. The extracellular domain of the CAR derives from a monoclonal antibody directed against the tumoral target. This extracellular domain consists of a single-chain variable fragment (scFv), which is the result of combining the variable heavy (VH) and the variable light (VL) chain by a linker. The scFv domain is anchored to the transmembrane region/domain by an aminoacidic hinge or spacer (Figure 1).
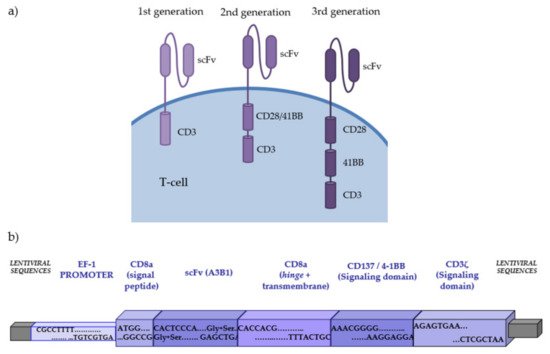
Figure 1. (a) Different generations of CAR-T cells. First-generation CAR-T cells include an intracellular domain. Second-generation CAR-T cells incorporate an additional co-stimulatory domain. Third-generation CAR-T cells include multiple co-stimulatory domains. (b) Structure of second-generation anti-CD19 CAR developed at the Immunology Department of Hospital Clínic de Barcelona.
The activation of CAR-T cells is the result of the recognition of the antigen by the scFv region, which concludes in the clustering and immobilization of the CAR molecules [13]. Intracellularly, the co-stimulatory domain chosen (typically sequences of CD28 or 4-1BB co-stimulatory regions) will determine the intracellular signaling and also could influence CAR dynamics, with the presence of a 4-1BB co-stimulation domain conferring slower expansion and longer persistence compared with the presence of a CD28 co-stimulation domain, which leads to rapid expansion but less durability [14][15]. Increased proliferation and expansion have also been associated with the 4-1BB co-stimulation domain when compared with CD28 [16]. Additionally, other co-stimulatory domains such as OX40, CD27, and inducible T-cell co-stimulator (ICOS) have been tested [13].
Subsequently, tyrosine-based activation motifs (ITAM) domains on the CD3ζ chain are phosphorylated, fostering the intracellular signaling through ZAP70 protein [17], which promotes CAR proliferation, cytokine release, metabolic alterations, and cytotoxicity. The release of granules with perforin and granzyme is the main mechanism associated with the CAR antitumoral effect. However, other death receptors such as BH3-interacting domain death agonist (BID) and FAS-associated death domain protein (FADD) have also been associated with CAR antitumoral function [17][18][19].
The process to manufacture CAR-T can take several weeks and can be performed in different ways. In our experience with CAR-T production from an academic clinical trial with a locally developed anti-CD19 CAR-T (ARI-0001) [20], patients provide the starting material: their own lymphoapheresis products; subsequently, in our proposal, this apheresis product is subjected to CD4- and CD8-positive selection, cultured and activated using anti-CD3 and anti-CD28 antibodies, by using a bioreactor that manages all the process under a close system by a sterile tubing set. After 24 h of activation, T-cells are transduced using our CAR-containing lentivirus and maintained in culture for expansion with IL-7 and IL-15 along 9–12 days.
Anti-CD19 CAR T-cells have shown outstanding results in clinical trials against leukemia and lymphoma [14][15][21][22][23][24], inducing high rates of response in patients with relapsed/refractory (R/R) B-cell lymphoproliferative disorders. In B-ALL, CD19-CAR therapy has achieved 60–93% complete responses (CRs) across several studies (reviewed in [25]) (as explained in more depth below). In R/R non-Hodgkin lymphoma (NHL) patients, CD19-CAR therapy achieved 40–90% of CR in heavily pretreated patients as reported by some of the main clinical assay JULIET [26], ZUMA-1 [27], and TRANSCEND NHL 001 [28].
This led to the approval by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) of two CAR-T cell therapies, Tisagenlecleucel (Kymriah) for relapsed/refractory (R/R) B-cell acute lymphoblastic leukemia (ALL) and R/R diffuse large B-cell lymphoma (DLBCL), and Axicabtagene Ciloleucel (Yescarta) for R/R DLBCL, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. Recently, brexucabtagene autoleucel (Tecartus) for R/R mantle cell lymphoma and lisocabtagene maraleucel (BREYANZI) for DLBCL, high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B were approved by FDA as well. At a more local level, there is also our CART19 cell therapy, a fully academic CAR approved by the Spanish Agency of Medicines (AEMPS) (ARI-0001) for R/R ALL, approved under the European “Hospital Exemption” rule.
Cell-based immunotherapies have revolutionized the approach to treat cancer. One of the main advantages of this therapy is that CAR T-cells bind their tumor target antigen specifically and efficiently enough to eliminate cancer cells, in an HLA-independent manner [29]. This prevents the tumor escape mechanism of downregulating the expression of HLA to avoid T-cell immune surveillance. Unlike chemotherapy or radiotherapy, the specificity of this approach avoids the unnecessary killing of healthy cells and tissues. Furthermore, the CD19 target of the CAR19 covers most of the B-cell malignancies, over 95%, making it extremely versatile and useful. Finally, a big difference with conventional cancer treatments such as chemotherapy and radiation is that CAR-T is considered living drugs, capable of proliferating and remains for years in the patient [30]. Despite the outstanding results of the CAR19 therapy, there are some limitations that currently are impairing its general use. One of the most important is the availability and fitness of T-cells used to manufacture the CAR19, as patients heavily treated might suffer from T-cell lymphopenia, and that can prevent them from being eligible for this therapy. This issue could potentially be solved using modified allogeneic T-cells to avoid GVHD manufacturing the CAR-T. Another issue related to the patient’s condition is the time that currently is necessary to generate the CAR-T; some rapidly progressive patients might not be able to wait. In some cases, clinicians can perform bridge chemotherapy to lower the tumor burden temporarily.
Another disadvantage is the CAR-T cell-associated toxicities such as cytokine release syndrome (CRS) and the immune effector cell-associated neurotoxicity syndrome (ICANS), both clinically manageable. Finally, another big hurdle is the tumor escape by target antigen loss, relapse patients with CD19 negative disease have been described in several clinical trials [31]. Strategies to overcome this problem include targeting multiple antigens with a bispecific or a tandem CAR [32][33].
Although clinical trials testing CAR-T targeting CD22 or BCMA are also presenting encouraging results for B-ALL [34] and multiple myeloma [35], and additional information will also arrive soon from the recently FDA-approved CAR-BCMA [36][37], in this review, we focus on the already approved anti-CD19+ CAR T-cell therapies.
3. Autologous or Allogenic CAR-T
Currently, there is an active area of investigation regarding the importance of the origin of the manufactured CAR T-cells. One approach is to obtain T-cells from the recipient’s allo-HSCT donor (allogenic) and the other one from the patient itself (autologous) (Figure 2).
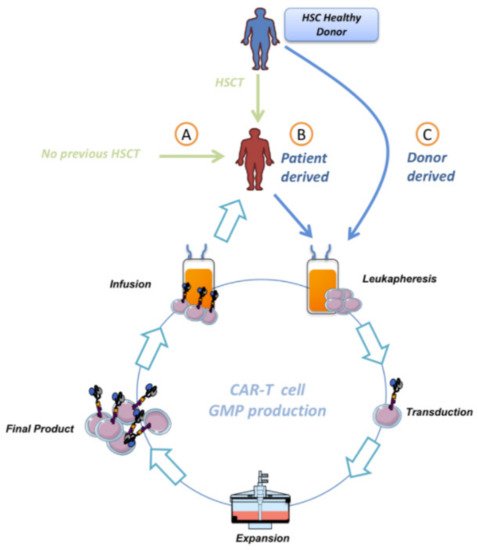
Figure 2. Different protocols in CAR-T therapy for B-cell malignancies. T-cells from patients with B-cell malignancies could be obtained from patients after HSCT (B) or without previous HSCT (A). Further, T-cells could be obtained from each recipient’s allo-HSCT healthy donor (C).
One of the requirements of using CAR-T derived from the HSC’s donor is the complete HSC engraftment before leukapheresis. This clearly begs the question of whether this CAR-T product should be considered allogeneic or autologous. Complete engraftment implies that the receptor’s hematopoietic system should be considered equal to donor’s, so one of the main issues of allogeneic CARTs such as alloimmunization (rejection of allo-CAR-T by receptor’s immune system) could be avoided, providing a major benefit from this procedure. Currently, CAR T-cells are considered autologous, independently whether the patient has received or not an allo-HSCT and the level of chimerism of the T-cells in the transplanted patients; however, from the biological point of view, CAR-T could only be considered autologous if the T-cells are from a patient that did not receive a previous allo-HSCT. For all the other possible scenarios, we should be talking about allogenic CART cells, where T-cells can be sourced from the same donor as the allo-HSCT, or directly from the allo-HSCT transplanted patient. The origin of T-cells used to manufacture the CAR-T has an outstanding impact on the overall process, from differences in the biological activity to potentially different costs in production. For instance, T-cells obtained from a heavily treated patient might be limited in numbers, and that can affect the administered dose, as well as the possibility to reinfuse CAR T-cells in case of necessity. Moreover, the manufacturing process is longer than using T-cells from a healthy donor, and more prone to failure. This represents an important issue for patients that show disease progression before CAR T-cells are available [38]. On the other hand, T-cells from healthy donors, even though not limited in numbers, could cause GVHD or be rapidly eliminated by the receptor’s immune system [38][39]. In any case, the use of T-cells in patients after allo-HSCT for manufacturing CAR-T opens an additional aspect of controversy. As commented, when T-cells are obtained and manufactured from the transplanted patients who will receive the infusion, these are considered autologous, even when these cells are genetically identical to the donor of allo-HSCT. In fact, in this situation, it is often possible to have access to the donor to obtain healthy T-cells. Unexpectedly, if the CAR-T development is made from newly obtained cells from the donor, this product will be considered by drug agencies an allogenic advanced therapy medicinal product (ATMP), different from the autologous ATMP developed from the transplanted patients. This aspect is very relevant because the current regulatory interpretation prevents the use of “better” cells from healthy donors who never received treatment previously and could provide functionally better CAR T-cells for the patients (or at least with lower accumulative cell potential injuries from previous treatments received by the patients).
Several clinical trials have compared CAR T-cells sourced from patients who received or did not receive an allo-HSCT. These studies achieved a similar minimal-residual disease and complete remission between groups [23][40]. In addition, there were no differences in treatment-related adverse events such as cytokine release syndrome and neurotoxicity. Although these clinical trials reported similar clinical outcomes, it is less common to use T-cells sourced from a patient who did not receive an allo-HSCT. As stated, allo-HSCT is one of the first standard treatments for B-cell malignancies, but we should consider that not all patients have the option of an HLA-compatible stem cell donor, or this procedure is not recommended for their clinical condition.
Numerous studies have reported that relapsed allo-HSCT patients with B cell malignancies could benefit from post-HSCT CAR-T infusion from the original HSC donor. Brudno et al. [41] conducted a clinical trial where CAR T-cells, targeting CD19, were obtained from each recipient’s allo-HSCT donor. This could lead to a major advantage as these cells have not been subjected to previous therapies administered to the patient to diminish the tumoral burden, but that could also diminish its numbers or function. According to the authors, no chemotherapy or other therapies were administered due to concern that the introduction of CAR T-cells into a recipient with depleted lymphocytes might cause severe GVHD. Eight of twenty treated patients obtained remission, which included six complete remissions and two partial remissions. The response rate was highest for acute lymphoblastic leukemia, with four of five patients obtaining minimal residual disease-negative complete remission. Responses also occurred in chronic lymphocytic leukemia and lymphoma. It is especially relevant to the fact that no acute GVHD was reported, suggesting that this strategy could reinforce the GVT effect without increasing the risk of GVHD. These results are in line with previous studies that reported regression of B-cell malignancies resistant to standard donor lymphocyte infusions without causing GVHD [42][43].
Focusing on the T-cells sourced from transplanted patient, there are still some challenges for manufacturing CARTs because the biological characteristics of these T-cells could negatively impact by the previous lines of treatment [38]. Moreover, T-cells could be dysfunctionally associated with immunosuppression treatment derived from transplants. Further, there are no data about chimerism before manufacturing CAR-T from a transplanted patient. Considering the percentage of chimerism could be important for having an idea about the T-cell fitness and indirectly associate it with a better or worse outcome with the manufacturing CARTs (transduction and expansion of CAR T-cells).
In summary, a chimerism analysis from the leukapheresis product would give us the ability to better compare clinical results between CAR-T generated from allo-HSC transplanted patients and CAR T-cells produced from matching HSCT-donor, trying to better define in an easy way the best product for each patient. “Starting material” for any product should be mainly defined by genetic characteristics, ahead of the extraction site (donor o patient) considerations. The final success of CAR-T therapy is obviously dependent on this origin, above general normative considerations.
References
- Padala, S.A.; Kallam, A. Diffuse Large B Cell Lymphoma; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276.
- Jacobson, C.A.; Longo, D.L. Chapter 104: Non-Hodgkin’ s Lymphoma. In Harrison’s Principles of Internal Medicine; McGraw Hill Education: New York, NY, USA, 2021; pp. 1–22.
- Lynegar, V.; Shimanovsky, A. Leukemia; StatPearls Publishing: Treasure Island, FL, USA, 2020.
- Patel, K.; Pagel, J.M. Current and future treatment strategies in chronic lymphocytic leukemia. J. Hematol. Oncol. 2021, 14, 1–20.
- Saadeh, S.S.; Litzow, M.R. Hematopoietic stem cell transplant in adults with acute lymphoblastic leukemia: The present state. Expert Rev. Hematol. 2018, 11, 195–207.
- Özgür Yurttaş, N.; Eşkazan, A.E. Novel therapeutic approaches in chronic myeloid leukemia. Leuk. Res. 2020, 91, 106337.
- Blazar, B.R.; Hill, G.R.; Murphy, W.J. Dissecting the biology of allogeneic HSCT to enhance the GvT effect whilst minimizing GvHD. Nat. Rev. Clin. Oncol. 2020, 17, 475–492.
- Liu, J.; Zhong, J.F.; Zhang, X.; Zhang, C. Allogeneic CD19-CAR-T cell infusion after allogeneic hematopoietic stem cell transplantation in B cell malignancies. J. Hematol. Oncol. 2017, 10, 1–8.
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355.
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 6230.
- Rivat, C.; Santilli, G.; Gaspar, H.B.; Thrasher, A.J. Gene Therapy for Primary Immunodeficiencies. Hum. Gene Ther. 2012, 23, 668–675.
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544.
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448.
- Zhang, H.; Snyder, K.M.; Suhoski, M.M.; Maus, M.V.; Kapoor, V.; June, C.H.; Mackall, C.L. 4-1BB Is Superior to CD28 Costimulation for Generating CD8+Cytotoxic Lymphocytes for Adoptive Immunotherapy. J. Immunol. 2007, 179, 4910–4918.
- Ramello, M.C.; Benzaïd, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabón-Saldaña, M.; Darville, L.; Fang, B.; Rix, U.; et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12, eaap9777.
- Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283.
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567.
- Castella, M.; Boronat, A.; Martín-Ibáñez, R.; Rodríguez, V.; Suñé, G.; Caballero, M.; Marzal, B.; Pérez-Amill, L.; Martín-Antonio, B.; Castaño, J.; et al. Development of a Novel Anti-CD19 Chimeric Antigen Receptor: A Paradigm for an Affordable CAR T Cell Production at Academic Institutions. Mol. Ther. Methods Clin. Dev. 2019, 12, 134–144.
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38.
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554.
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459.
- Ortíz-Maldonado, V.; Rives, S.; Castellà, M.; Alonso-Saladrigues, A.; Benítez-Ribas, D.; Caballero-Baños, M.; Baumann, T.; Cid, J.; Garcia-Rey, E.; Llanos, C.; et al. CART19-BE-01: A Multicenter Trial of ARI-0001 Cell Therapy in Patients with CD19+ Relapsed/Refractory Malignancies. Mol. Ther. 2021, 29, 636–644.
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355.
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56.
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42.
- Abramson, M.J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, D.M.A.; Wang, M.L.; Arnason, J.E.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, M.C.; et al. Pivotal Safety and Efficacy Results from Transcend NHL 001, a Multicenter Phase 1 Study of Lisocabtagene Maraleucel (liso-cel) in Relapsed/Refractory (R/R) Large B Cell Lymphomas. Blood 2019, 134, 241.
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398.
- Cappell, K.M.; Sherry, R.M.; Yang, J.C.; Goff, S.L.; Vanasse, D.A.; McIntyre, L.; Rosenberg, S.A.; Kochenderfer, J.N. Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J. Clin. Oncol. 2020, 38, 3805–3815.
- Ruella, M.; Maus, M.V. Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput. Struct. Biotechnol. J. 2016, 14, 357–362.
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Liu, Y.; Zhang, W.; Wang, C.; Zhang, Y.; et al. Bispecific Chimeric Antigen Receptor Targeting Both CD19 and CD22 T Cell Therapy in Adults with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia. SSRN Electron. J. 2020, 8, 1–10.
- Tong, C.; Zhang, Y.; Liu, Y.; Ji, X.; Zhang, W.-Y.; Guo, Y.; Han, X.; Ti, D.; Dai, H.; Wang, C.; et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B cell lymphoma. Blood 2020, 136, 1632–1644.
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-CAR T Cells Induce Remissions in CD19-CAR Naïve and Resistant B-ALL. Nat. Med. 2018, 24, 20–28.
- Rodríguez-Lobato, L.G.; Ganzetti, M.; De Larrea, C.F.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1243.
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324.
- Usmani, S.Z.; Berdeja, J.G.; Madduri, D.; Jakubowiak, A.J.; Agha, M.E.; Cohen, A.D.; Hari, P.; Yeh, T.-M.; Olyslager, Y.; Banerjee, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen (BCMA)-directed chimeric antigen receptor T-cell (CAR-T) therapy, in relapsed/refractory multiple myeloma (R/R MM): Updated results from CARTITUDE-1. J. Clin. Oncol. 2021, 39, 8005.
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199.
- Dinofia, A.K.; Grupp, S.A. Will allogenic CAR T cells for CD19+ malignances take autologous CAR T cells ‘off the shelf’? Nat. Rev. Clin. Oncol. 2021, 18, 195–196.
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331.
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121.
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139.
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973, Erratum in 2014, 123, 3364.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
694
Revisions:
2 times
(View History)
Update Date:
26 Oct 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No