 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | James R Bamburg | + 4510 word(s) | 4510 | 2021-10-19 05:33:52 | | | |
| 2 | Jason Zhu | -49 word(s) | 4461 | 2021-10-21 04:09:48 | | | | |
| 3 | Jason Zhu | -35 word(s) | 4426 | 2021-10-25 03:57:51 | | | | |
| 4 | Jason Zhu | -35 word(s) | 4426 | 2021-10-25 04:00:09 | | |
Video Upload Options
Proteins of the actin depolymerizing factor (ADF)/cofilin family are ubiquitous among eukaryotes and are essential regulators of actin dynamics and function. Mammalian neurons express cofilin-1 as the major isoform, but ADF and cofilin-2 are also expressed. All isoforms bind preferentially and cooperatively along ADP-subunits in F-actin, affecting the filament helical rotation, and when either alone or when enhanced by other proteins, promotes filament severing and subunit turnover.
1. Introduction
The importance of the ADF/cofilin family of proteins with respect to the regulation of actin dynamics in virtually every eukaryotic cell in every kingdom and phyla has resulted in numerous reviews covering most aspects of these proteins, especially within the nervous system where they have essential functions in synaptic plasticity associated with memory and learning [1][2][3]. With so much known about these proteins, why is another review useful at this time? Within the past few years, new post-translational modifications of these proteins have been reported, some of which are isoform specific and, thus, potentially provide independent regulatory pathways.
2. Actin Dynamics and ADF/Cofilin Basics
Actin assembly will occur spontaneously in vitro when monomers are above a critical concentration and ionic conditions allow them to self-associate to form a nucleating trimer. Assembly can occur with either ATP-actin or ADP-actin. Two parallel strands of subunits with the same polarity assemble in a helical structure to produce a filament (F-actin). If ADP-actin is assembled, the filament formed is an equilibrium polymer with the same critical (equilibrium) concentration at each end. Filament ends are denoted as barbed or pointed, nomenclature taken from the arrowhead decoration of F-actin by proteolytic fragments of myosin. If assembly is initiated with ATP-actin, the hydrolysis of ATP to ADP-Pi is rapid (~2 s) with a much slower loss of inorganic phosphate (minutes when measured in vitro), the latter being accompanied by a change in the filament structure resulting in different on/off rate constants (and, thus, different equilibrium/critical concentrations) at the slower growing ADP-actin pointed end [4]. Many F-actin binding proteins, including ADF/cofilin, can influence the dissociation rate of Pi [5]. The subunits of ATP-actin continue to add rapidly to the barbed end, but the loss of subunits from the pointed end occurs as the actin monomer pool declines. At a steady state, ATP-actin subunits continue to add to the barbed end, whereas ADP-actin subunits are lost from the pointed end to maintain a constant ratio of filament mass to monomers, but the subunits will continue to treadmill through a filament if ATP is available ( Figure 1 ). In vivo, monomer sequestering proteins prevent spontaneous filament nucleation, so growth occurs from either severed existing filaments or from specific nucleation factors, such as formins or the Arp2/3 complex discussed below.
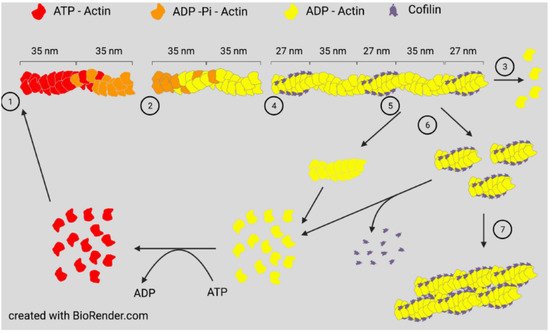
All three proteins, ADF, cofilin-1, and cofilin-2, are expressed in adult mammalian neurons [14] and bind both globular (G)-actin and filamentous (F)-actin with a strong preference (>40 fold) for binding ADP-actin [15][16]. At low concentrations with respect to actin subunits (1:750), cofilin is an effective F-actin severing protein. At higher concentrations, severing efficiency declines but occurs at the junction between cofilin-saturated and unsaturated regions, which are sites of increased strain [12][17], but stable cofilin-actin complexes can also nucleate growth [9][16] ( Figure 1 ).
There are subtle differences in how mammalian ADF and cofilin-1 modulate actin, and although each can rescue many functions of the other in cell behavioral assays, there are some isoform-specific functions [18]; these could arise because of differences in the protein’s inherent activity or reflect differences in their sites of translation or cellular regulation. Inherent activity differences are observed in the assembly of ADF-actin and cofilin-actin complexes, each of which assembles into filaments that appear identical to F-actin saturated with cofilin or ADF, but the critical concentration for assembly of ADF-actin is >5 μM, whereas it is ~1 μM for cofilin-actin, which is approximately the same as for ADP-actin alone. Thus, ADF-actin may contribute to the intracellular pool of monomer, whereas cofilin-actin will not [11][16][19]. For similar reasons, cofilin-actin filaments might survive longer in cells where stress has resulted in a reduction in ATP and filaments approach their equilibrium state.
The question of how expression of ADF and cofilin is controlled is also of great interest. During myoblast differentiation to myocytes in chickens, which only express ADF and cofilin-2, there is a big switch in expression to cofilin-2 as the myocytes mature [20]. There is also a big change in the expression of the skeletal muscle α-actin isoform and in the assembled actin pool, raising the question “is ADF or cofilin expression tied to the monomeric actin pool?” This was addressed in mouse C2C12 myoblasts by expressing a mutant form of actin that could not assemble into filaments. We previously proposed that ADF expression but not that of cofilin is downregulated by a post-transcriptional mechanism and the expression of total actin is also controlled [21]. This feedback control is modulated by the actin monomer pool as depolymerizing actin filaments using latrunculin A also decreases ADF but not cofilin expression. ADF, cofilin, and various actin isoforms are known transcriptional targets of the SRF and its cofactor MKL1 [22]. The activation of MKL1/SRF gene transcription is closely coupled to free cellular G-actin levels [23], suggesting an alternate mechanism for linkage between ADF expression and the actin monomer pool. Differential regulation of individual SRF target genes through miRNAs or competing repressive factors may contribute to distinctive ADF vs. cofilin expression mediated by SRF [24].
3. Actin Dynamics in Neuritogenesis and Neurite Growth
Brain development is morphologically normal in ADF null (ADF KO) mice [25], but cofilin is required for all aspects of neuronal development. Neuritogenesis, the first morphological steps in neuronal differentiation, does not occur in neuroblasts of ADF KO mice in which cofilin-1 is conditionally knocked out [26]. Although not measured specifically in neuronal progenitor cells, cofilin-2, which is highly upregulated in muscle at later stages of development, is probably not expressed or expressed at very low levels at this stage [27]. What is striking from this study is the presence of membrane parallel actin filaments in electron micrographs of the neuroblasts, suggesting that the formation of neurites is regulated by the need for cofilin-induced disassembly of the membrane proximal F-actin, such as what has been described for polarizing fibroblasts [28]. The breakdown of the membrane proximal F-actin by cofilin is followed by nucleated assembly of actin filaments perpendicular to the membrane, driving filopodial and lamellipodial protrusions that emerge as the GC.
De novo neurite outgrowth and regeneration in adult neurons also requires actin turnover mediated by cofilin [14]. Two cofilin activities can be distinguished based on site directed mutants: S94D can depolymerize but not sever filaments and Y82F can sever but not enhance depolymerization [29][30]. When these mutants are expressed individually in an adult axon regeneration system in which endogenous ADF and cofilin are silenced, severing is the essential activity for regeneration [14].
Although the outgrowth and migration of the GC is reasonably well understood at the molecular level [31], the question of how cofilin regulation fits into the guidance of growth cones has been more controversial. Ratio imaging of immunofluorescence images of stained total cofilin to phospho-ADF/cofilin in GCs showed higher levels of active cofilin along the region of the growth cone opposite to the side in contact with a repulsive guidance cue (aggrecan) [32]. In chick retinal or dorsal root ganglion neurons, which express ADF and not cofilin-1, an attractive turning response was observed to a gradient of a membrane permeable source of either WT or S3A ADF, but not S3E, with increased filament barbed ends on the growth cone side turning toward the source of the ADF [33], suggesting that enhanced severing and generation of new barbed ends might be sufficient for setting the direction of protrusion and growth.
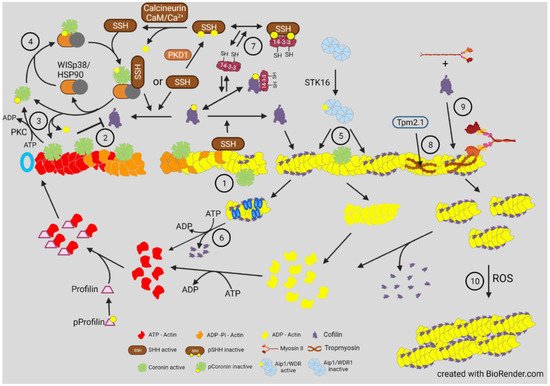
However, opposite results for phospho-cofilin distribution were observed in Xenopus laevis spinal neurons responding in culture to a gradient of bone morphogenic protein 7 (BMP7), an attractant for these growth cones in their early developmental stage (0–8 h) [34]. During this period, the BMP7/receptor activated LIMK1 was essential in turning and resulted in increased phospho-cofilin on the growth cone side in the direction of turning. Surprisingly, the response to BMP-7 reversed in neurons between 8 and 20 h in culture, and BMP7 became a repulsive cue due to expression of a transient receptor potential channel (TRPC) that responded to BMP7 by allowing calcium influx, which activated calcineurin, a Ca2+ -calmodulin dependent phosphatase previously shown to activate SSH in neurons [35][34] ( Figure 4 ). The distribution of cofilin activity across the growth cone was reversed during repulsion, which was blocked with calcineurin inhibitors. The timing of the switch between attraction and repulsion in vitro correlates with ventral projections of commissural neurons in vivo, which first benefitted from the attraction to the BMP7 producing cells and then by repulsion that aids in their growth past the site of BMP7 release. Taken together, these findings suggest that it is more than just a bias in activated cofilin that is required for the dynamic regulation of actin. It is possible that a decrease in activated cofilin might enhance its severing activity, which is optimal at about 10 nM on isolated filaments in vitro [8]. However, as shown in the bead experiment described in Figure 2 , there is likely a broad range of cofilin concentrations (nM to μM) that can support steady state dynamics of actin [36]. In cells, cofilin phosphocycling might be more important than the amount of cofilin that is in the dephosphorylated pool. Alternatively, other proteins that enhance severing and turnover of cofilin-bound actin discussed above ( Figure 3 ) may also have gradients of activity across a growth cone and perhaps may not require any changes in phosphorylated cofilin to control turning response.
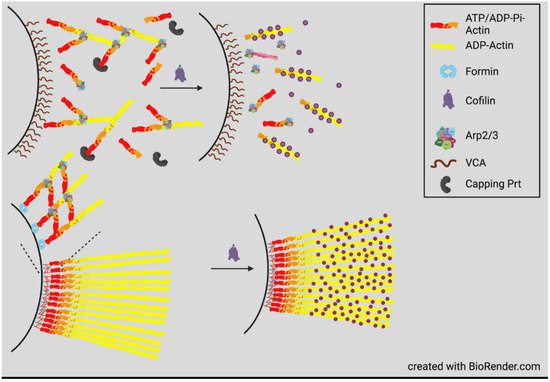
4. Actin Dynamics in Neurite Consolidation and Branching
Temporal regulation of actin dynamics is also important in the consolidation process by which the neurite shaft forms behind the extending growth cone and in neurite branching. A current theory of consolidation is that it requires repressing protrusive activity stimulated by cortactin, an activator of the Arp2/3 complex, that helps form branched actin networks associated with new protrusions [49]. Cortactin is also an enhancer of deposition of extracellular matrix material stimulating branching [50]. Cortactin is highly sensitive to degradation by calpain, a calcium-activated protease within neurite shafts. By degrading cortactin, calpain limits new protrusions. The branching of more mature neurites probably occurs by reversal of this pathway through inhibition of calpain by phosphorylation catalyzed by the cyclic AMP-dependent protein kinase A (PKA). This pathway likely explains the branching at sites of contact between neurites and neurotrophin-coated beads that locally activate adenylate cyclase to produce cAMP [51]. Neurite branching also requires septins, a family of GTP-binding proteins that can assemble into filaments, rings, and mesh works and which then recruit cortactin to new sites of collateral branch formation [52]. Cortactin also aids septins in directing neurite microtubules toward the site of protrusion where branched actin networks assemble on the tip of the microtubule through the activity of the adenomatous polyposis coli (APC) protein [53]. The penetration of a protrusion by a microtubule is required for the delivery of mitochondria and the development of the GC for neurite branch elongation [52][54].
Significant branching of primary neurites also occurs during early stages of outgrowth in cultured hippocampal neurons. The consolidation phase of the neurite at the base of the growth cone is often broken by the transit of new lamellipodial-like processes (waves), usually starting at the soma and migrating along the neurite [55][56]. During the period of axonogenesis, waves increase in frequency along the future axon [57][58]. On some substrates, neurites elongate faster when the wave reaches the growth cone, although myosin II-induced alterations in growth cone shape may result in an apparent surge forward [59]. During early neurite extension in cultured hippocampal neurons, waves move into and expand the tip of a filopodium and form a new growth cone establishing a branch off the neurite. The ability of the waves to extend down the neurite depends upon their binding to substrate through receptors, such as the L1-cell adhesion molecule (L1-CAM), which couples with cortactin and the F-actin network that is undergoing retrograde flow (treadmilling) with the forward protrusion of the membrane driven by actin assembly. This “molecular clutch” is provided by shootin-1b [60]. Actomyosin and the microtubule motor dynein also contribute to the forces for neurite elongation [61]. Waves transport actin toward the neurite tip probably by a preferential reutilization of actin subunits disassembled by cofilin from treadmilling filaments and is, to date, one of only two demonstrable mechanisms for neurite actin transport, the other being similar with nucleating hot spots that result in a biased anterograde elongation of filaments within neurites [62].
Long-term stabilization of the neurite shaft comes from membrane associated actin-rings that are spaced at about 190 nm by spectrin tetramers [63][64]. Although initially thought to be short, capped filaments, recent studies using platinum replicate electron microscopy and super-resolution microscopy show that filaments in the rings are composed of two long intertwined F-actins connected by a dense meshwork of aligned spectrins [65]. The rings form earlier in a presumptive axon than in a dendrite and extend throughout the axon shaft, whereas they are not as complete throughout its length in a dendrite [66]. Rings compartmentalize the membrane and restrict diffusion of lipids within the axon initial segment [67], but they do not form early enough to explain neurite consolidation. Rings associate with myosin II, which has a scaffolding and/or contractile role that can alter axon electrophysiology [68]. The actin ring network in axons is more stable with respect to remodeling than in dendrites [69][70], but the rings can expand and contract during cargo passage. Although the effects of cofilin on ring stability and turnover have yet to be reported, it is possible that cofilin on its own may not directly interact with the actin in rings given their structure and extensive spectrin cross-linking and myoII binding.
If cofilin is unable to disassemble ring actin, what other proteins might do so? Calpain, the cortactin-degrading protein in shaft consolidation, also degrades ring structures during a Ca2+ -dependent degenerative response [71], although a milder Ca2+ -induced F-actin disassembly might be provided by gelsolin, a Ca2+ -dependent severing and barbed-end capping protein that is expressed in neurons [72]. Alternatively, an F-actin severing activity has been identified as a function of a large (250 kDa) multidomain protein containing a leucine-rich repeat and kinase domains called Lrrk2. As a monomer or dimer, Lrrk2 severs F-actin in vitro but loses this activity when further oligomerized [73]. Its severing activity is of interest because mutations in Lrrk2 that affect severing are among the most common mutations found in familial Parkinson’s disease (PD). Furthermore, Lrrk2 interacts with α-synuclein, resulting in Lrrk2 oligomerization, loss of severing activity, and enhanced stability of F-actin in Drosophila models of PD. These can be partially reversed in flies by overexpressing Drosophila cofilin (twinstar). The decreased turnover of F-actin in neurons with mutant Lrrk2 results in the mis-localization of dynamin-related protein 1 (Drp1) and subsequent mitochondrial elongation and dysfunction. The role of α-synuclein, the major amyloid component of Lewy bodies, in dysregulating Lrrk2 is of particular interest for understanding dementia, which occurs in a significant number of PD patients who have triplication of the α-synuclein gene [74].
5. Actin Dynamics in Nuclear and Mitochondrial Function
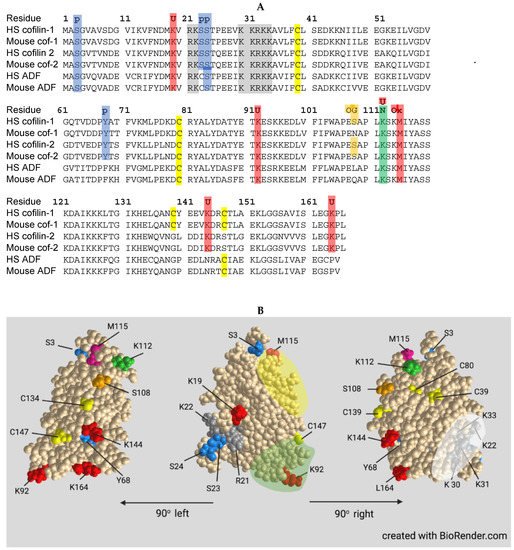
Cofilin contains a nuclear localization sequence similar to that of the SV40 large T-antigen with additional residues comprising a bipartite NLS more recently identified (Figure 4). All three ADF/cofilin isoforms have been identified in stress-induced rod-shaped cofilin-actin bundles (nuclear rods) from different cell types. Cofilin transports a complex of monomeric actin and importin 9 into the nucleus via the nuclear pore complex (NPC). Actin export from the nucleus through the NPC is mediated by profilin and exportin 6. The dynamics of nuclear F-actin are mediated at least in part by a pool of nuclear cofilin, which increases with enhanced transport of G-actin. Monomeric nuclear actin fills many roles in transcriptional regulation, first suggested in 1984, including serving as a component of all three RNA polymerases and in many chromatin-remodeling complexes. Alterations in nuclear transport of actin has major effects on responses to growth stimulatory pathways, such as that activated by serum response factor, and on the overall transcriptome.
A cofilin-regulated transient pool of nuclear F-actin is required for chromatin decondensation following cell exit from mitosis into G1. The transient F-actin pool assembles at the nuclear envelope and results in protrusions and nuclear volume expansion as well as in chromatin organization in daughter nuclei. Nuclear cofilin-1 inactivation by phosphorylation occurs concomitantly with transition into G1 as nuclear F-actin increases. However, nuclear actin assembly promoted by G-protein coupled receptor (GPCR)-mediated calcium signaling via the formin INF2 also enhances nuclear protrusions and volume expansion in G1 that require filament bundling by alpha-actinin 4, a mediator of endothelial mesenchymal transformation, tumorigenesis, and cancer metastasis. Significantly regarding their intranuclear functions, alpha-actinins 2 and 4 are found as components of intranuclear rods that are devoid of cofilin, but which form in response to the expression of a skeletal muscle actin mutation (V163L) that causes intranuclear rod myopathy, suggesting that rod formation is a conserved mechanism for sequestering potentially harmful proteins and is part of the actin stress response.
Mitochondria, often major sources of ROS production in stressed cells, can undergo both fission and fusion to modulate their numbers and cellular distribution; abnormalities in their dynamics and transport are linked to many neurodegenerative diseases. The fission and fusion processes allow the mixing of mitochondrial contents. Removal of damaged mitochondria by mitophagy is also a normal process in healthy cells, but mitochondria also serve as a sensor to trigger cell death through apoptosis. Actin reorganization for both mitochondrial fission, requiring Drp1, and mitophagy to remove non-functional mitochondria, are dependent on actin reorganization by cofilin. Some apoptotic signaling pathways, especially those in which oxidative stress is the trigger, involved oxidation of cofilin and its targeting to the mitochondrion where through interactions with cyclase associated protein1 (CAP1) it leads to release of apoptotic factors.
6. Cofilin-Actin Rods in Neurodegenerative Disorders
The formation of cytoplasmic rod-shaped bundles containing cofilin and actin havebeen studied in neurons: (1) overexpressing or acutely activating (dephosphorylating) cofilin; (2) exposed to energetic, oxidative or excitotoxic stress including hypoxia/ischemia; and (3) exposed to factors associated with progressive cognitive decline in age-related neurodegenerative diseases. Rods form within neurites, but only rarely in the cell soma, in response to energetic and oxidative stress. Rods induced by energy depletion have been isolated from both cortical neurons and non-neuronal cells and contain the two cytoplasmic actin isoforms and ADF/cofilin in a ratio between total actin and total ADF/cofilin of 1:1. ADF and cofilin in rods are present at about their same expression level as within the cell type from which they are isolated. Rods are sensitive to non-ionic detergents which should not be used for cell permeabilization if immunostaining of coflin in rods is desired. Most cofilin in rods occurs as a cofilin dimer cross-linked by a disulfide bond between C39 and C147.
The rapidity of rod formation in energetically stressed and glutamate-treated neurons and in brain slices undergoing oxygen deprivation resulted in studies on rod formation in ischemic brain injury (stroke). Four different mouse models of brain ischemia were examined, each initiating rod formation but with differing patterns and in all models rods formed exclusively in neuronal processes. Photothrombic lesions produced the most localized ischemic event; rods started forming within 1 h and reached their maximum at 24 h, localizing within the distal edge of the infarct region and slightly into the surrounding non-ischemic tissue but never within the core. In rats subjected to MCAO and reperfusion, regions with rods showed almost complete disappearance of functional mitochondria, and synaptic activity declined in the neurons with rods. Treatments that promoted cofilin phosphorylation reduced rod formation whereas enhancing cofilin dephosphorylation increased rods and exacerbated synapse loss. Overexpressing the cofilin inactivating kinase, LIMK1, reduced rod formation, protected synapses, and attenuated cofilin-mediated apoptosis, increasing neuronal survival following stroke, supporting the finding that decreased cofilin activity is beneficial in recovery from stroke.
Rods are associated with several cognitive disorders and share a common pathway for their formation in the subpopulation of neurons affected. Rod formation in culture systems is causative of synaptic deficits, but rods only form in a subset of hippocampal neurons in response to disease-related degenerative factors. However, only a subpopulation of neurons in the hippocampus undergo loss of dendritic spines in a sleep deprivation model in which cognitive deficits are significant. Given the importance of cofilin in modulating dynamic actin processes for synaptic function, it is not yet possible to conclude that rods per se are causative with respect to the cognitive dysfunctions that arise.
Although rods from energetically stressed cells have been isolated and characterized, components of rods that develop during neurodegenerative disease progression, other than actin and cofilin, are unknown. Since rod formation induced by the PrPC/NOX pathway is in a small subpopulation of neurons and fewer rods form per neuron than during energetic stress, their isolation for compositional analysis is challenging. One approach might be to use rod reporters to visualize the rod and laser capture microdissection coupled to mass spectrometry for their compositional analysis. Certainly, much work lies ahead to obtain a complete understanding of the many beneficial and detrimental activities of this interesting ADF/cofilin protein family.
References
- Kang, D.E.; Woo, J.A. Cofilin, a master node regulating cytoskeletal pathogenesis in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, S131–S144.
- Hoffmann, L.; Rust, M.B.; Culmsee, C. Actin(g) on Mitochondria—A role for cofilin1 in neuronal cell death pathways. Biol. Chem. 2019, 400, 1089–1097.
- Ben Zablah, Y.; Merovitch, N.; Jia, Z. The Role of ADF/cofilin in synaptic physiology and Alzheimer’s disease. Front. Cell Dev. Biol. 2020, 8, 594998.
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576.
- Blanchoin, L.; Pollard, T.D. Mechanism of Interaction of Acanthamoeba Actophorin (ADF/Cofilin) with Actin Filaments. J. Biol. Chem. 1999, 274, 15538–15546.
- McGough, A.; Pope, B.; Chiu, W.; Weeds, A. Cofilin changes the twist of f-actin: Implications for actin filament dynamics and cellular function. J. Cell Biol. 1997, 138, 771–781.
- Galkin, V.E.; Orlova, A.; Lukoyanova, N.; Wriggers, W.; Egelman, E.H. Actin depolymerizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. J. Cell Biol. 2001, 153, 75–86.
- Andrianantoandro, E.; Pollard, T.D. Mechanism of Actin Filament Turnover by Severing and Nucleation at Different Concentrations of ADF/Cofilin. Mol. Cell 2006, 24, 13–23.
- Koffer, A.; Edgar, A.J.; Bamburg, J.R. Identification of Two Species of Actin Depolymerizing Factor in Cultures of BHK Cells. J. Muscle Res. Cell Motil. 1988, 9, 320–328.
- Hayden, S.M.; Miller, P.S.; Brauweiler, A.; Bamburg, J.R. Analysis of the Interactions of Actin Depolymerizing Factor with G- and F-Actin. Biochemistry 1993, 32, 9994–10004.
- Yeoh, S.; Pope, B.; Mannherz, H.G.; Weeds, A. Determining the Differences in Actin Binding by Human ADF and Cofilin. J. Mol. Biol. 2002, 315, 911–925.
- Suarez, C.; Roland, J.; Boujemaa-Paterski, R.; Kang, H.; McCullough, B.R.; Reymann, A.-C.; Guérin, C.; Martiel, J.-L.; De la Cruz, E.M.; Blanchoin, L. Cofilin Tunes the Nucleotide State of Actin Filaments and Severs at Bare and Decorated Segment Boundaries. Curr. Biol. 2011, 21, 862–868.
- Flores, L.R.; Keeling, M.C.; Zhang, X.; Sliogeryte, K.; Gavara, N. Lifeact-GFP Alters F-Actin Organization, Cellular Morphology and Biophysical Behaviour. Sci. Rep. 2019, 9, 3241.
- Tedeschi, A.; Dupraz, S.; Curcio, M.; Laskowski, C.J.; Schaffran, B.; Flynn, K.C.; Santos, T.E.; Stern, S.; Hilton, B.J.; Larson, M.J.E.; et al. ADF/Cofilin-Mediated Actin Turnover Promotes Axon Regeneration in the Adult CNS. Neuron 2019, 103, 1073–1085.e6.
- Ressad, F.; Didry, D.; Xia, G.X.; Hong, Y.; Chua, N.H.; Pantaloni, D.; Carlier, M.F. Kinetic Analysis of the Interaction of Actin-Depolymerizing Factor (ADF)/Cofilin with G- and F-Actins. Comparison of Plant and Human ADFs and Effect of Phosphorylation. J. Biol. Chem. 1998, 273, 20894–20902.
- Chen, H.; Bernstein, B.W.; Sneider, J.M.; Boyle, J.A.; Minamide, L.S.; Bamburg, J.R. In Vitro Activity Differences between Proteins of the ADF/Cofilin Family Define Two Distinct Subgroups. Biochemistry 2004, 43, 7127–7142.
- Hocky, G.M.; Sindelar, C.V.; Cao, W.; Voth, G.A.; De La Cruz, E.M. Structural Basis of Fast- and Slow-Severing Actin-Cofilactin Boundaries. J. Biol. Chem. 2021, 296, 100337.
- Tahtamouni, L.H.; Shaw, A.E.; Hasan, M.H.; Yasin, S.R.; Bamburg, J.R. Non-Overlapping Activities of ADF and Cofilin-1 during the Migration of Metastatic Breast Tumor Cells. BMC Cell Biol. 2013, 14, 45.
- Devineni, N.; Minamide, L.S.; Niu, M.; Safer, D.; Verma, R.; Bamburg, J.R.; Nachmias, V.T. A Quantitative Analysis of G-Actin Binding Proteins and the G-Actin Pool in Developing Chick Brain. Brain Res. 1999, 823, 129–140.
- Morgan, T.E.; Lockerbie, R.O.; Minamide, L.S.; Browning, M.D.; Bamburg, J.R. Isolation and Characterization of a Regulated Form of Actin Depolymerizing Factor. J. Cell Biol. 1993, 122, 623–633.
- Minamide, L.S.; Painter, W.B.; Schevzov, G.; Gunning, P.; Bamburg, J.R. Differential Regulation of Actin Depolymerizing Factor and Cofilin in Response to Alterations in the Actin Monomer Pool. J. Biol. Chem. 1997, 272, 8303–8309.
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-Actin Signaling to the MRTF Coactivators Dominates the Immediate Transcriptional Response to Serum in Fibroblasts. Genes Dev. 2014, 28, 943–958.
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin Dynamics Control SRF Activity by Regulation of Its Coactivator MAL. Cell 2003, 113, 329–342.
- Kawakami-Schulz, S.V.; Verdoni, A.M.; Sattler, S.G.; Jessen, E.; Kao, W.W.-Y.; Ikeda, A.; Ikeda, S. Serum Response Factor: Positive and Negative Regulation of an Epithelial Gene Expression Network in the Destrin Mutant Cornea. Physiol. Genom. 2014, 46, 277–289.
- Ikeda, S.; Cunningham, L.A.; Boggess, D.; Hawes, N.; Hobson, C.D.; Sundberg, J.P.; Naggert, J.K.; Smith, R.S.; Nishina, P.M. Aberrant Actin Cytoskeleton Leads to Accelerated Proliferation of Corneal Epithelial Cells in Mice Deficient for Destrin (Actin Depolymerizing Factor). Hum. Mol. Genet. 2003, 12, 1029–1037.
- Flynn, K.C.; Hellal, F.; Neukirchen, D.; Jacob, S.; Tahirovic, S.; Dupraz, S.; Stern, S.; Garvalov, B.K.; Gurniak, C.; Shaw, A.E.; et al. ADF/cofilin-mediated actin retrograde flow directs neurite formation in the developing brain. Neuron 2012, 76, 1091–1107.
- Gurniak, C.B.; Perlas, E.; Witke, W. The Actin Depolymerizing Factor N-Cofilin Is Essential for Neural Tube Morphogenesis and Neural Crest Cell Migration. Dev. Biol. 2005, 278, 231–241.
- Bisaria, A.; Hayer, A.; Garbett, D.; Cohen, D.; Meyer, T. Membrane-Proximal F-Actin Restricts Local Membrane Protrusions and Directs Cell Migration. Science 2020, 368, 1205–1210.
- Moriyama, K.; Yahara, I. Two activities of cofilin, severing and accelerating directional depolymerization of actin filaments, are affected differentially by mutations around the actin-binding helix. EMBO J. 1999, 18, 6752–6761.
- Moriyama, K.; Yahara, I. The actin-severing activity of cofilin is exerted by the interplay of three distinct sites on cofilin and essential for cell viability. Biochem. J. 2002, 365, 147–155.
- Leite, S.C.; Pinto-Costa, R.; Sousa, M.M. Actin Dynamics in the Growth Cone: A Key Player in Axon Regeneration. Curr. Opin. Neurobiol. 2021, 69, 11–18.
- Flynn, K.C.; Pak, C.W.; Bamburg, J.R. Regulation of Growth Cone Initiation and Actin Dynamics by ADF/cofilin. In Intracellular Mechanisms for Neuritogenesis; Springer: New York, NY, USA, 2007; pp. 25–56. ISBN 0-387-33128-X.
- Marsick, B.M.; Flynn, K.C.; Santiago-Medina, M.; Bamburg, J.R.; Letourneau, P.C. Activation of ADF/Cofilin Mediates Attractive Growth Cone Turning toward Nerve Growth Factor and Netrin-1. Dev. Neurobiol. 2010, 70, 565–588.
- Wen, Z.; Han, L.; Bamburg, J.R.; Shim, S.; Ming, G.; Zheng, J.Q. BMP Gradients Steer Nerve Growth Cones by a Balancing Act of LIM Kinase and Slingshot Phosphatase on ADF/Cofilin. J. Cell Biol. 2007, 178, 107–119.
- Wang, Y.; Shibasaki, F.; Mizuno, K. Calcium Signal-Induced Cofilin Dephosphorylation Is Mediated by Slingshot via Calcineurin. J. Biol. Chem. 2005, 280, 12683–12689.
- Bleicher, P.; Sciortino, A.; Bausch, A.R. The Dynamics of Actin Network Turnover Is Self-Organized by a Growth-Depletion Feedback. Sci. Rep. 2020, 10, 6215.
- Gandhi, M.; Achard, V.; Blanchoin, L.; Goode, B.L. Coronin switches roles in actin disassembly depending on the nucleotide state of actin. Mol. Cell 2009, 34, 364–374.
- BoseDasgupta, S.; Moes, S.; Jenoe, P.; Pieters, J. Cytokine-induced macropinocytosis in macrophages is regulated by 14-3-3ζ through its interaction with serine-phosphorylated coronin 1. FEBS J. 2015, 282, 1167–1181.
- Cai, L.; Marshall, T.W.; Uetrecht, A.C.; Schafer, D.A.; Bear, J.E. Coronin 1B Coordinates Arp2/3 Complex and Cofilin Activities at the Leading Edge. Cell 2007, 128, 915–929.
- Howell, M.; Brickner, H.; Delorme-Walker, V.D.; Choi, J.; Saffin, J.-M.; Miller, D.; Panopoulos, A.; DerMardirossian, C.; Fotedar, A.; Margolis, R.L.; et al. WISp39 Binds Phosphorylated Coronin 1B to Regulate Arp2/3 Localization and Cofilin-Dependent Motility. J. Cell Biol. 2015, 208, 961–974.
- Chan, K.T.; Creed, S.J.; Bear, J.E. Unraveling the Enigma: Progress towards understanding the coronin family of actin regulators. Trends Cell Biol. 2011, 21, 481–488.
- Ge, P.; Durer, Z.A.O.; Kudryashov, D.; Zhou, Z.H.; Reisler, E. Cryo-EM reveals different coronin binding modes for ADP- and ADP-BeFx Actin Filaments. Nat. Struct. Mol. Biol. 2014, 21, 1075–1081.
- Gohla, A.; Bokoch, G.M. 14-3-3 Regulates Actin Dynamics by Stabilizing Phosphorylated Cofilin. Curr. Biol. 2002, 12, 1704–1710.
- Mizuno, K. Signaling Mechanisms and Functional Roles of Cofilin Phosphorylation and Dephosphorylation. Cell Signal. 2013, 25, 457–469.
- Gateva, G.; Kremneva, E.; Reindl, T.; Kotila, T.; Kogan, K.; Gressin, L.; Gunning, P.W.; Manstein, D.J.; Michelot, A.; Lappalainen, P. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr Biol. 2017, 27, 705–713.
- Chua, B.T.; Volbracht, C.; Tan, K.O.; Li, R.; Yu, V.C.; Li, P. Mitochondrial Translocation of Cofilin Is an Early Step in Apoptosis Induction. Nat. Cell Biol. 2003, 5, 1083–1089.
- Klamt, F.; Zdanov, S.; Levine, R.L.; Pariser, A.; Zhang, Y.; Zhang, B.; Yu, L.-R.; Veenstra, T.D.; Shacter, E. Oxidant-Induced Apoptosis Is Mediated by Oxidation of the Actin-Regulatory Protein Cofilin. Nat. Cell Biol. 2009, 11, 1241–1246.
- Bernstein, B.W.; Shaw, A.E.; Minamide, L.S.; Pak, C.W.; Bamburg, J.R. Incorporation of Cofilin into Rods Depends on Disulfide Intermolecular Bonds: Implications for Actin Regulation and Neurodegenerative Disease. J. Neurosci. 2012, 32, 6670–6681.
- Yang, C.; Svitkina, T. Filopodia Initiation: Focus on the Arp2/3 Complex and Formins. Cell Adh. Migr. 2011, 5, 402–408.
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell functions of a multifaceted actin-binding protein. Trends Cell Biol. 2018, 28, 79–98.
- Spillane, M.; Ketschek, A.; Donnelly, C.J.; Pacheco, A.; Twiss, J.L.; Gallo, G. Nerve growth factor-induced formation of axonal filopodia and collateral branches involves the intra-axonal synthesis of regulators of the actin-nucleating Arp2/3 Complex. J. Neurosci. 2012, 32, 17671–17689.
- Hu, J.; Bai, X.; Bowen, J.R.; Dolat, L.; Korobova, F.; Yu, W.; Baas, P.W.; Svitkina, T.; Gallo, G.; Spiliotis, E.T. Septin-driven coordination of actin and microtubule remodeling regulates the collateral branching of axons. Curr. Biol. 2012, 22, 1109–1115.
- Efimova, N.; Yang, C.; Chia, J.X.; Li, N.; Lengner, C.J.; Neufeld, K.L.; Svitkina, T.M. Branched actin networks are assembled on microtubules by adenomatous polyposis coli for targeted membrane protrusion. J. Cell Biol. 2020, 219, e202003091.
- Armijo-Weingart, L.; Gallo, G. It takes a village to raise a branch: Cellular mechanisms of the initiation of axon collateral branches. Mol. Cell. Neurosci. 2017, 84, 36–47.
- Ruthel, G.; Banker, G. Actin-dependent anterograde movement of growth-cone-like structures along growing hippocampal axons: A novel form of axonal transport? Cell Motil. Cytoskeleton 1998, 40, 160–173.
- Ruthel, G.; Banker, G. Role of moving growth cone-like “wave” structures in the outgrowth of cultured hippocampal axons and dendrites. J. Neurobiol. 1999, 39, 97–106.
- Flynn, K.C.; Pak, C.W.; Shaw, A.E.; Bradke, F.; Bamburg, J.R. Growth cone-like waves transport actin and promote axonogenesis and neurite branching. Dev. Neurobiol. 2009, 69, 761–779.
- Winans, A.M.; Collins, S.R.; Meyer, T. Waves of actin and microtubule polymerization drive microtubule-based transport and neurite growth before single axon formation. Elife 2016, 5, e12387.
- Mortal, S.; Iseppon, F.; Perissinotto, A.; D’Este, E.; Cojoc, D.; Napolitano, L.M.R.; Torre, V. Actin waves do not boost neurite outgrowth in the early stages of neuron maturation. Front. Cell. Neurosci. 2017, 11, 402.
- Minegishi, T.; Uesugi, Y.; Kaneko, N.; Yoshida, W.; Sawamoto, K.; Inagaki, N. Shootin1b mediates a mechanical clutch to produce force for neuronal migration. Cell Rep. 2018, 25, 624–639.e6.
- Minegishi, T.; Inagaki, N. Forces to drive neuronal migration steps. Front. Cell Dev. Biol. 2020, 8, 863.
- Chakrabarty, N.; Dubey, P.; Tang, Y.; Ganguly, A.; Ladt, K.; Leterrier, C.; Jung, P.; Roy, S. Processive flow by biased polymerization mediates the slow axonal transport of actin. J. Cell Biol. 2019, 218, 112–124.
- Xu, K.; Zhong, G.; Zhuang, X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 2013, 339, 452–456.
- Unsain, N.; Stefani, F.D.; Cáceres, A. The actin/spectrin membrane-associated periodic skeleton in neurons. Front. Synaptic Neurosci. 2018, 10, 10.
- Vassilopoulos, S.; Gibaud, S.; Jimenez, A.; Caillol, G.; Leterrier, C. Ultrastructure of the axonal periodic scaffold reveals a braid-like organization of actin rings. Nat. Commun. 2019, 10, 5803.
- Han, B.; Zhou, R.; Xia, C.; Zhuang, X. Structural organization of the actin-spectrin-based membrane skeleton in dendrites and soma of neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E6678–E6685.
- Albrecht, D.; Winterflood, C.M.; Sadeghi, M.; Tschager, T.; Noé, F.; Ewers, H. Nanoscopic compartmentalization of membrane protein motion at the axon initial segment. J. Cell Biol. 2016, 215, 37–46.
- Costa, A.R.; Sousa, S.C.; Pinto-Costa, R.; Mateus, J.C.; Lopes, C.D.; Costa, A.C.; Rosa, D.; Machado, D.; Pajuelo, L.; Wang, X.; et al. The membrane periodic skeleton is an actomyosin network that regulates axonal diameter and conduction. Elife 2020, 9, e55471.
- Lavoie-Cardinal, F.; Bilodeau, A.; Lemieux, M.; Gardner, M.-A.; Wiesner, T.; Laramée, G.; Gagné, C.; De Koninck, P. Neuronal activity remodels the f-actin based submembrane lattice in dendrites but not axons of hippocampal neurons. Sci. Rep. 2020, 10, 11960.
- Leterrier, C. Putting the Axonal Periodic Scaffold in Order. Curr. Opin. Neurobiol. 2021, 69, 33–40.
- Zhou, R.; Han, B.; Xia, C.; Zhuang, X. Membrane-associated periodic skeleton is a signaling platform for RTK transactivation in neurons. Science 2019, 365, 929–934.
- González-Tapia, D.; González-Tapia, D.C.; Vázquez-Hernández, N.; Martínez-Torres, N.I.; Flores-Soto, M.; González-Burgos, I. Modifications to cytoskeleton-associated proteins in dendritic spines underlie the adaptive plasticity involved in long term reference memory. Neurobiol. Learn. Mem. 2020, 172, 107247.
- Sarkar, S.; Bardai, F.; Olsen, A.L.; Lohr, K.M.; Zhang, Y.-Y.; Feany, M.B. Oligomerization of Lrrk Controls actin severing and α-synuclein neurotoxicity in vivo. Mol. Neurodegener. 2021, 16, 33.
- Halliday, G.M.; Leverenz, J.B.; Schneider, J.S.; Adler, C.H. The neurobiological basis of cognitive impairment in Parkinson’s Disease. Mov. Disord. 2014, 29, 634–650.

