 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniela Elena Ilie | + 1830 word(s) | 1830 | 2021-09-29 08:17:07 | | | |
| 2 | Rita Xu | Meta information modification | 1830 | 2021-10-19 10:50:04 | | | | |
| 3 | Rita Xu | Meta information modification | 1830 | 2021-10-19 10:51:16 | | |
Video Upload Options
Mastitis is one of the most frequently encountered diseases in dairy cattle, negatively affecting animal welfare and milk production. For this reason, contributions to understanding its genomic architecture are of great interest. Genome-wide association studies (GWAS) have identified multiple loci associated with somatic cell score (SCS) and mastitis in cattle.
1. Introduction
2. Animals and Phenotypes
3. Principal Component Analysis

4. Significant SNPs Associated with SCS
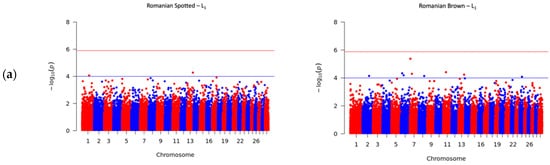
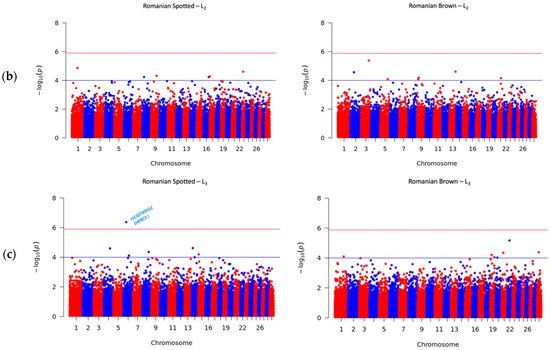
A total number of 41 SNPs were detected in the RS and RB breeds, of which 40 SNPs passed the suggestive threshold and, ultimately, one SNP passed the significance threshold after Bonferroni correction and was associated with SCS in the RS breed in L3 (Figure 2c, left side). For the RS breed, 3 out of 15 SNPs were located near three known genes, and one SNP overlapped the HERC3 gene. Of the fifteen SNPs in the RS breed, AX-106761943 (rs110749552) was located on chromosome 6 within the HERC3 gene (HECT and RLD domain containing E3 ubiquitin protein ligase 3) and was the most significant SNP for SCS (log10 (p) = 6.37). In the RB breed, the genome-wide analysis detected 26 SNPs that reached genome-wide significance for association with SCS. Among these SNPs, 14 out of 26 SNPs were located near 12 known genes; two SNPs, AX-106741653 and AX-115114947, were located near the AKAP8 gene, and two other SNPs, AX-106735825 (rs43585636) and AX- 117085949 (rs43585209) were located near the COL12A1 gene, respectively. None of the 15 detected SNPs in the RS breed were in common with the detected 26 SNPs that reached genome-wide significance for association with SCS in the RB breed.
This study revealed that different markers and candidate genes were found for the two breeds in our study. In addition, all identified SNPs have been distinct among the three parities, denoting that SCS is influenced by different genes according to parity. The different sets of markers discovered in this work compared to studies reported in the literature can be attributed to various factors. We can assume that the power of detecting SNPs can be lower in the RB breed compared to the RS breed as a consequence of the comparatively lower number of genotyped RB animals used in the analysis. Furthermore, previously known mastitis related genes such as LUZP2, AKAP8 and MEGF10 were also identified in our study as candidate genes for SCS in the RB breed. For this specific case in the RB breed, further studies using larger sample sizes should be performed in order to validate previous results. Finally, an additional factor that may influence the different sets of candidate SNPs/breed is the distinct genetic backgrounds of the two breeds as they have different levels of clustering, as shown in previous studies on Romanian cattle breeds.
The specific novel SNPs and candidate genes located around them reported in the present study can be considered as candidates involved in SCS and mastitis resistance; how- ever, these need to be kept in perspective, and polymorphisms in those genes should be fur- ther analyzed to highlight whether they influence the ability of dairy cows to resist mastitis.
5. Conclusions
Our study represents the first GWAS for SCS in Romanian dairy cattle and, thus, provides new perspective into the genetic architecture of udder infections in these native dairy cattle. We identified 41 SNPs and detected their significant associations with variation in SCS in dairy cattle. In both breeds, the SNPs and position of association signals were distinct among the three parities, denoting that mastitis is influenced by different genes according to parity. The results contribute to an increase in knowledge regarding the proportion of genetic variability explained by SNPs for SCS in dairy cattle and, particularly, in Romanian native cattle. However, further large-scale studies of a vast number of native dairy cattle breeds are needed in order to investigate others markers and genes that could be involved in mastitis.
References
- Rupp, R.; Boichard, D. Genetics of resistance to mastitis in dairy cattle. Vet. Res. 2003, 34, 671–688.
- Aarestrup, F.M.; Jensen, N.E.; Ostergård, H. Analysis of associations between major histocompatibility complex (BoLA) class I haplotypes and subclinical mastitis of dairy cows. J. Dairy Sci. 1995, 78, 1684–1692.
- Dietz, A.B.; Cohen, N.D.; Timms, L.; Kehrli, M.E., Jr. Bovine lymphocyte antigen class II alleles as risk factors for high somatic cell counts in milk of lactating dairy cows. J. Dairy Sci. 1997, 80, 406–412.
- Kelm, S.C.; Detilleux, J.C.; Freeman, A.E.; Kehrli, M.E., Jr.; Dietz, A.B.; Fox, L.K.; Butler, J.E.; Kasckovics, I.; Kelley, D.H. Genetic association between parameters of innate immunity and measures of mastitis in periparturient Holstein cattle. J. Dairy Sci. 1997, 80, 1767–1775.
- Gilliespie, B.E.; Jayarao, B.M.; Dowlen, H.H.; Oliver, S.P. Analysis and frequency of bovine lymphocyte antigen DRB3.2 alleles in Jersey cows. J. Dairy Sci. 1999, 82, 2049–2053.
- Takeshima, S.; Nakai, Y.; Ohta, M.; Aida, Y. Characterization of DRB3 alleles in the MHC of Japanese Shorthorn Cattle by polymerase chain reaction-sequence-based typing. J. Dairy Sci. 2002, 85, 1630–1632.
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; Van Tassell, C.P.; Sonstegard, T.S.; et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10, R42.
- Meredith, B.K.; Kearney, F.J.; Finlay, E.K.; Bradley, D.G.; Fahey, A.G.; Berry, D.P.; Lynn, D.J. Genome-wide associations for milk production and somatic cell score in Holstein-Friesian cattle in Ireland. BMC Genet. 2012, 13, 21.
- Zhou, J.; Liu, L.; Chen, C.J.; Zhang, M.; Lu, X.; Zhang, Z.; Huang, X.; Shi, Y. Genome-wide association study of milk and reproductive traits in dual-purpose Xinjiang Brown cattle. BMC Genom. 2019, 20, 827.
- Meredith, B.K.; Berry, D.P.; Kearney, F.; Finlay, E.K.; Fahey, A.G.; Bradley, D.G.; Lynn, D.J. A genome-wide association study for somatic cell score using the Illumina high-density bovine beadchip identifies several novel QTL potentially related to mastitis susceptibility. Front. Genet. 2013, 4, 229.
- Strillacci, M.G.; Frigo, E.; Schiavini, F.; Samoré, A.B.; Canavesi, F.; Vevey, M.; Cozzi, M.C.; Soller, M.; Lipkin, E.; Bagnato, A. Genome-wide association study for somatic cell score in Valdostana Red Pied cattle breed using pooled DNA. BMC Genet. 2014, 15, 106.
- Nani, J.P.; Raschia, M.A.; Poli, M.A.; Calvinho, L.F.; Amadio, A.F. Genome-wide association study for somatic cell score in Argentinean dairy cattle. Livest. Sci. 2015, 175, 1–9.
- The Animal Quantitative Trait Loci (QTL) Database. Available online: https://www.animalgenome.org/cgi-bin/QTLdb/BT/index (accessed on 20 May 2021).
- Wiggans, G.R.; Shook, G.E. A lactation measure of somatic-cell count. J. Dairy Sci. 1987, 70, 2666–2672.
- Interbull. Code of Practice. Available online: https://interbull.org/ib/codeofpractice (accessed on 25 August 2021).
- Parker Gaddis, K.L.; Null, D.J.; Cole, J.B. Explorations in genome-wide association studies and network analyses with dairy cattle fertility traits. J. Dairy Sci. 2016, 99, 6420–6435.
- Welderufael, B.G.; Løvendahl, P.; de Koning, D.J.; Janss, L.L.G.; Fikse, W.F. Genome-Wide Association Study for Susceptibility to and Recoverability from Mastitis in Danish Holstein Cows. Front. Genet. 2018, 9, 141.


