 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kerstin Borgmann | + 2905 word(s) | 2905 | 2021-09-30 10:21:33 | | | |
| 2 | Catherine Yang | Meta information modification | 2905 | 2021-10-19 03:18:26 | | |
Video Upload Options
Cancer stem cells (CSCs) are a tumor cell population maintaining tumor growth and promoting tumor relapse if not wholly eradicated during treatment. CSCs are often equipped with molecular mechanisms making them resistant to conventional anti-cancer therapies whose curative potential depends on DNA damage-induced cell death.
1. Introduction
Recent discoveries for cancer therapy, such as antibody-based immunotherapy, and various predictive biomarkers, have shifted the focus from standard uniform treatment towards personalized approaches [1][2]. Although significant improvement in life expectancy has been reached for certain tumors, tumor relapses still represent a major threat for patients with metastatic disease [3][4][5]. Mounting evidence suggests that recurrences can be attributed, at least in part, to the existence of a pluripotent subpopulation of tumor cells. The cancer stem cells (CSCs) possess the ability to self-renew and exhibit an enhanced therapy resistance. Therefore, they can re-populate a tumor after initially successful therapy [5][6][7][8]. Since their first isolation from acute myeloid leukemia by the group of John Dick, CSCs have been identified in various solid tumors [9][10]. As with their non-malignant stem-cell counterparts, CSCs are equipped with various mechanisms to protect their genome from endogenous or treatment-induced damage [6][11].
Less DNA damage in CSCs as compared to their non-CSC counterparts was reported in several studies that quantified DNA damage after genotoxic therapies or at baseline levels. This quantification was made, e.g., using residual γH2A.X foci analysis (for example, more efficient foci resolution was shown for lung cancer CD133 + cells [12], murine cancer CD29 + CD24 high cells [13], head and neck squamous cell carcinoma (HNSCC) ALDH + cells [14], glioblastoma CSC populations in patient-derived cell lines [15]) or by comet assay (for example the lower amount of DNA damage was found in glioblastoma patient-derived CD133 + cells [16], CD133 + CD44 + colon cancer cells [17] and murine cancer CD29 + CD24 high cells [13]). However, some studies showed no difference in the levels of DNA damage quantified in the same way in CSCs and non-CSC populations [18][19][20]. This controversy can be partially attributed to DNA repair pathways that can be activated independently on H2AX phosphorylation [21], different timepoints used for these analyses, inconsistent methods for CSC isolation or enrichment, and, in some cases, lack of CSC functional validation. At the same time, quantification of DNA damage and DNA repair response does not always correlate with tumor resistance to the DNA-damaging treatment [19][20][22].
Interestingly, while the accumulation of mutations is a major threat for the maintenance of embryonic and adult stem-cell populations, a certain level of genomic instability can promote malignant transformation and CSC induction [23][24]. According to the unified model of tumor evolution suggested by Kreso and Dick, CSCs are located at the apex of the tumor cell hierarchy [8]. Yet, the ongoing process of tumor evolution enables CSCs to acquire additional mutations that further enhances their tumorigenic potential and therapy resistance [8]. Hereditary mutations in key DNA repair genes, such as the breast cancer susceptibility (BRCA) and Fanconi anemia (FA) genes, drastically emphasize the role of genomic instability as a driving force in tumorigenesis [23][25][26]. Importantly, previous reports suggest that this relationship is not linear. Long-term accumulation of DNA damage can lead to permanent replication stress and intolerable mutation burden, eventually activating tumor suppression mechanisms such as apoptosis and senescence [27][28][29]. In this context, suppression of the DNA damage response (DDR) and its associated signaling might increase the DNA damage tolerance and prolongs the survival of CSCs [20][22][30]. However, various studies demonstrated that CSCs crucially rely on the up-regulation of DNA repair pathways to counteract the adverse effects of genomic instability, as reviewed in detail elsewhere [6][11][31]. Consequently, the high basal levels of cell cycle checkpoint kinases and DNA repair proteins provide CSCs with a robust armor against genotoxic treatment. Yet, the dependency on a balanced DDR also provides promising opportunities for targeted therapy, as demonstrated for poly(ADP-ribose) polymerase 1 (PARP1) inhibitors in BRCA-mutant tumors [32].
Despite the obvious link between DNA repair pathways and the CSC phenotype, the mechanisms mediating and modulating this crosstalk remain only partially understood. Notably, the double-edged role of the DDR in tumor evolution and therapy resistance suggests a complex relationship that goes beyond the elimination of treatment-induced DNA damage.
2. The Role of DNA Repair Proteins in the CSC Induction and Maintenance
Despite the emergence of targeted therapies and immunomodulatory approaches, DNA-damaging agents such as ionizing irradiation and platinum-based chemotherapeutics are still a primary strategy for the treatment of various cancers [33][34]. Thus, many studies investigated the expression and activity of DDR factors as determinants of resistance towards these therapies [35][36][37]. Interestingly, an increasing number of reports demonstrate that alterations in DNA repair gene expression equally modulate the cancer cell self-renewal capability, in vivo tumorigenicity, and invasiveness—characteristic properties of CSCs. At first glance, the connection between DNA repair and the cellular stemness program might not be obvious. Yet, accumulating evidence suggests that DNA repair genes possess many regulatory functions in addition to their canonical role in the DNA repair process. This chapter describes selected examples of stemness regulation via co-regulation of transcription, interference with CSC-related signaling, and ROS detoxification and discusses the implications of DNA repair gene expression modulation for the CSC phenotype.
Thus, the corporation with these transcription factors enables DNA repair proteins to influence the CSC cellular program at the apex of the regulation hierarchy.
It is well known that deficiencies in DNA repair pathways, such as the FANC/BRCA pathway or the DNA mismatch repair (MMR), predispose to cancer due to increased genomic instability. However, the overexpression of DNA repair genes is also often associated with malignant transformation [38][39][40][41][42]. A rapidly growing body of evidence suggests that the altered expression of DNA proteins promotes EMT, metastasis, and de-differentiation of tumor cells by interfering with major CSC-related signaling pathways. Due to the numerous reports of possible interactions, the following paragraphs focus on a few well-investigated examples.
In summary, the described interactions of DNA repair proteins with various CSC-related signaling pathways support a scenario in which functional tumor-suppressive crosstalk requires fine-tuned protein expression, precise posttranslational modification, and coordinated sub-cellular localization of all involved factors. Furthermore, overexpression, inactivation as well as loss-of-function or gain-of-function mutations on both sides can potentially disbalance the regulatory network, leading to malignant transformation, de-differentiation, and therapy resistance.
3. The Role of DNA Repair Signaling in the Immune Response against Cancer Cells
Besides the impact of the DDR on the maintenance and plasticity of CSCs, direct and indirect effects of the DDR can modulate antitumor immunity. Importantly, damaged DNA and impaired chromosome segregation during mitosis lead to accumulation of genomic DNA in the cytosol. For example, the DDR-induced release of nuclear DNA into the cytoplasm in prostate cancer cells was dependent on the endonuclease methyl methanesulphonate and ultraviolet-sensitive 81 (MUS81), which cleaves DNA upon stalling of replication forks [43]. Although low levels of cytosolic DNA are usually degraded by DNases such as three prime repair exonuclease 1 (TREX1), an accumulation of nuclear DNA in the cytoplasm can stimulate various DNA-sensing pattern recognition receptors (PRRs). For example, the RNA polymerase III recognizes AT-rich dsDNA and activates retinoic acid inducible gene I (RIG-I), which induces type I interferon (IFN) and the NF-κB pathway [44]. Furthermore, cyclic GMP-AMP synthase (cGAS) also detects cytosolic DNA and produces the second messenger cyclic GMP-AMP (cGAMP), resulting in the activation of stimulator of interferon genes (STING) and subsequent type I IFN production [45]. STING phosphorylation by TANK-binding kinase-1 (TBK1) results in the recruitment of interferon response transcription factor (IRF)3, which is involved in the signaling of type I IFN responses. The TBK1-mediated phosphorylation of IRF3 induces its dimerization, nuclear translocation, and transcription of target genes [46]. By promoting the antigen-presenting capacity of dendritic cells and stimulating the cytotoxic activity of CD8+ T cells and natural killer (NK) cells, type I IFNs can essentially contribute to antitumor immunity [47]. As STING is a key factor of the immune response to DNA damage, it has been shown that many tumors down-regulate STING or exhibit mutations, which leads to a dampened inflammatory response [48]. Recently, Suter et al. demonstrated that the inhibition of the cGAS-STING axis is at least partially mediated by interleukin (IL) 6 and downstream JAK2/STAT3 signaling [49]. Interestingly, IL-6 and STAT3 are key players in the generation and maintenance of CSCs, suggesting a mechanistical link [50]. Besides a type I IFN response, the sensing of cytoplasmic DNA via absent in melanoma 2 (AIM2) facilitates inflammasome formation and generation of IL-1β [51][52].
In addition to cytosolic DNA stimulating classical PRRs, certain molecules involved in the DDR closely interact with the immune system. For example, the DNA repair protein DNA-dependent protein kinase (DNA-PK) prolongs the half-life of the activated form of the transcription factor IRF3, which is involved in the signaling of type I IFN responses [46][53]. Furthermore, DNA-damaging agents induce an ATM-dependent small ubiquitin-like modifier (SUMO) modification of NF-κB essential modulator (NEMO), which fosters IκB kinase complex (IKK) and subsequent NF-κB activation [54]. Although a temporary inflammatory response may promote tumor elimination by activation and recruitment of immune cells, chronic inflammation can in turn lead to further DNA damage, for example by phagocyte-derived ROS, and thus foster tumor progression [55]. Importantly, the DDR also prevents the accumulation of additional cytosolic DNA and the subsequent triggering of cytosolic DNA sensors. As shown by Wolf et al., the DDR proteins RAD51 and replication protein A (RPA) prevent short ssDNA from passing the nuclear membrane and a knockdown of both molecules resulted in a cGAS-dependent activation of the type I IFN response [56]. Depletion of RAD51 is associated with accumulation of cytosolic DNA attributed to excessive degradation of the reverse replication fork by MRE11 exonuclease and consequent activation of STING signaling [57]. Interestingly, RAD51 was shown to be up-regulated in CSCs and drive the resistance of CSCs towards PARP1 inhibitors in TNBC and glioma [58][59] , Figure 1. Similarly, the loss of BRCA2 leads to cytosolic DNA accumulation and subsequent cGAS/STING-dependent up-regulation of IFN-stimulating genes [60]. Another study demonstrated that radiation delivered to tumor cells at a high dose in a single fraction induces expression of TREX1 DNA exonuclease leading to the accumulation of cytosolic DNA and activation of cGAS/STING signaling, illustrating the mechanism of radiation-induced innate immune signaling [61]. Furthermore, DNA damage in the S-phase of the cell cycle highly activates STING/TBK1/IRF3 signaling activation and expression of PD-L1 in breast cancer cells that lack functional FA/BRCA-signaling S-phase specific DNA repair mechanism [62].
Besides driving proinflammatory immune responses via STING and NF-κB, the DDR promotes tumor cells to express ligands for the activating NK cell receptors NKG2D (Killer cell lectin-like receptor subfamily K, member 1) and DNAM-1 (DNAX Accessory Molecule-1) via ATM and ATR signaling [79][80]. Functional experiments revealed that the aphidicolin-induced up-regulation of NKG2D ligands on T cell blasts increased their sensitivity towards IL-2-activated NK cells, which was partially reduced by addition of an anti-NKG2D antibody. These results are supported by Soriani et al., who showed that the degranulation of NK cells in the presence of myeloma cells was promoted upon treatment with doxorubicin or melphalan, while the addition of NKG2D and DNAM-1 blocking antibodies reduced the observed effect. Furthermore, a persistent DDR facilitates the senescence of tumor cells accompanied by the secretion of IL-6 and IL-8, which is also known as the senescence-associated secretory phenotype (SASP) [81]. Accumulating evidence suggests that the SASP promotes invasiveness, regenerative capacity and other characteristics associated with the generation of CSCs [82]. It has also been shown that DSBs induce PD-L1 expression by cancer cells via the ATM/ATR/CHK1 signaling axis and the IRF1 pathway [83][84][85]. Therefore, DDR-associated molecules emerged as potential biomarkers predicting the efficacy of checkpoint inhibitor (CPI) treatment. In a cohort of CPI-treated metastatic urothelial carcinoma, the activation of the DDR pathway was associated with a reduced TGF-β signaling as well as an increase in neoantigen load, activated immune cells, and survival time [86]. Another study found that mutations of the DDR pathway were associated with improved overall survival in a colorectal CPI-treated colorectal cancer (CRC) patient cohort, while there was no such correlation in the non-CPI cohort [87].
In conclusion, the DDR can modulate antitumor immunity by facilitating the production of proinflammatory cytokines and promoting tumor cells to express ligands for stimulating NK cell receptors. However, a functional DDR also prevents the accumulation of cytosolic DNA and subsequent activation of the immune system, which suggests particular implications for CSCs, as they frequently overexpress proteins involved in the DDR. Nevertheless, the effect of an altered DDR on the immune response towards CSCs remains to be discovered, as CSCs additionally create an immunosuppressive environment and exhibit various mechanisms to effectively evade the recognition and elimination by the immune system [88].
4. DNA Repair in Cancer Stem Cells as a Therapeutic Target
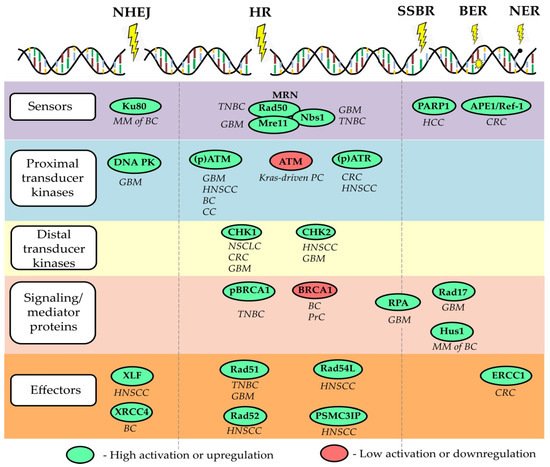
The fundamental properties of CSCs, such as unlimited self-renewal potential, differentiation capacity, and, consequently, tumor-maintaining properties, make them an ultimate therapeutic target for permanent tumor control. Thus, the utmost clinical importance of CSC is proven using CSC-related signatures as reliable prognostic biomarkers and by clinical trials aiming at targeting CSCs in different tumor entities. Targeting DNA repair in CSCs can be suggested as a promising anti-cancer therapeutic strategy as many preclinical studies revealed a high DNA repair capacity in CSCs. Indeed, CSCs in the different tumor entities are shown to be more proficient in the activation of the DDR signaling pathway due to the activation of its various components, including signal sensors, transducers, and effectors, as summarized in a recent review of Schultz et al. and shown in Figure 1 . On the other hand, defective DNA repair leads to genetic instability and cancer susceptibility [89]. Here, we will discuss the role of several DNA repair proteins as the most promising emerging targets to eliminate CSC populations and discuss the potential challenges of their clinical translation.
Inactivation of the p53 tumor suppressor is a hallmark of most types of cancer and one of the survival mechanisms for CSC with DNA repair deficiency [30][90]. Under DNA-damaging treatments, activation of p53 is required either for the cell cycle arrest to repair DNA lesions or for the induction of cell senescence and death if DNA damage is unrepairable. In the absence of p53-mediated G1- and G2/M-phase cell cycle arrest, cancer cells harboring DNA lesions are no longer arrested at the G1/S transition and progress through S-phase into the G2 phase and mitosis [91]. Thus, p53 plays a vital role in maintaining genome integrity, and its dysfunction leads to the accumulation of DNA mutations and genome instability. In response to UV treatment, impaired p53 activation in esophageal CSCs was associated with an attenuated G1 and G2/M-phase cell cycle arrest. Thus, it can be a potential mechanism of CSC survival upon DNA toxic therapy and cancer progression [30]. Furthermore, developing p53-activating therapeutic strategies including MDM2 and MDMX inhibition, induction of p53 transcription program by chemical compounds such as CP-31398 and PRIMA-1met, and synthetic lethality caused by p53 loss in combination with inhibition of protein kinases such as WEE1, FYN, and AURKA open further directions for targeting of CSCs deficient for p53 activation [92].
Enhanced DNA repair and extended cell cycle arrest upon DNA toxic treatments are not always a common feature of CSCs possessing relative radioresistance compared to their non-CSC counterparts [22][30]. Furthermore, up-regulation of a single or several DNA repair genes does not always correlate with more efficient DNA repair mechanisms. For example, a study based on the analysis of The Cancer Genome Atlas (TCGA) database showed that increased expression levels of a gene set, including 23 DNA repair genes, are associated with a deficient HR and inhibition of DNA repair machinery and therefore are indicative for the improved patients’ response to the DNA toxic therapies [93]. Another finding demonstrated that high expression of four DNA repair proteins such as Rif1, PARP-1 Binding Protein (PARI), RAD51, and Ku80 indicates low HR-dependent DNA repair efficiency and is associated with genomic instability and high sensitivity to platinum-based chemotherapy in NSCLC patients [94].
These studies are supported by a recent finding showing that high expression levels of single genes involved in HR-dependent DNA repairs such as POU5F1, PSMC3IP, and RAD54L are associated with better disease-free survival in patients with HNSCC. Furthermore, this study also showed that the transcription factor POU5F1 /Oct4 is a marker of HNSCC stem cells and acts as a transcriptional regulator of PSMC3IP and RAD54L expression, suggesting a mechanism of direct regulation of DNA repair by CSC-specific transcriptional factors [73]. A similar observation was also made for DNA repair genes representing the chromosomal (CIN) instability score in patients with HNSCC, indicating that high expression of individual DNA genes did not lead to the therapy resistance [95].
References
- Baumann, M.; Krause, M.; Overgaard, J.; Debus, J.; Bentzen, S.M.; Daartz, J.; Richter, C.; Zips, D.; Bortfeld, T. Radiation oncology in the era of precision medicine. Nat. Rev. Cancer 2016, 16, 234–249.
- Jackson, S.E.; Chester, J.D. Personalised cancer medicine. Int. J. Cancer 2015, 137, 262–266.
- Ganesh, K.; Massague, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44.
- Klein, C.A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 2020, 20, 681–694.
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711.
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862.
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24.
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291.
- Desai, A.; Yan, Y.; Gerson, S.L. Concise Reviews: Cancer Stem Cell Targeted Therapies: Toward Clinical Success. Stem Cells Transl. Med. 2019, 8, 75–81.
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648.
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA Damage in Stem Cells. Mol. Cell 2017, 66, 306–319.
- Desai, A.; Webb, B.; Gerson, S.L. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. 2014, 110, 538–545.
- Chang, C.H.; Zhang, M.; Rajapakshe, K.; Coarfa, C.; Edwards, D.; Huang, S.; Rosen, J.M. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Rep. 2015, 5, 378–391.
- Kurth, I.; Hein, L.; Mabert, K.; Peitzsch, C.; Koi, L.; Cojoc, M.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Cancer stem cell related markers of radioresistance in head and neck squamous cell carcinoma. Oncotarget 2015, 6, 34494–34509.
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203.
- Obara, E.A.A.; Aguilar-Morante, D.; Rasmussen, R.D.; Frias, A.; Vitting-Serup, K.; Lim, Y.C.; Elbaek, K.J.; Pedersen, H.; Vardouli, L.; Jensen, K.E.; et al. SPT6-driven error-free DNA repair safeguards genomic stability of glioblastoma cancer stem-like cells. Nat. Commun. 2020, 11, 4709.
- Peng, L.; Xiong, Y.; Wang, R.; Xiang, L.; Zhou, H.; Gu, H. Identification of a subpopulation of long-term tumor-initiating cells in colon cancer. Biosci. Rep. 2020, 40, BSR20200437.
- Karimi-Busheri, F.; Rasouli-Nia, A.; Mackey, J.R.; Weinfeld, M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast Cancer Res. 2010, 12, R31.
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Harding, A.; Kozlov, S.; Kijas, A.W.; Ensbey, K.S.; Walker, D.G.; Lavin, M.F. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Mol. Cancer Ther. 2012, 11, 1863–1872.
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392.
- Xie, A.; Puget, N.; Shim, I.; Odate, S.; Jarzyna, I.; Bassing, C.H.; Alt, F.W.; Scully, R. Control of sister chromatid recombination by histone H2AX. Mol. Cell 2004, 16, 1017–1025.
- Lundholm, L.; Haag, P.; Zong, D.; Juntti, T.; Mork, B.; Lewensohn, R.; Viktorsson, K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013, 4, e478.
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127.
- Lagasse, E. Cancer stem cells with genetic instability: The best vehicle with the best engine for cancer. Gene Ther. 2008, 15, 136–142.
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185.
- Wu, J.; Mu, Q.; Thiviyanathan, V.; Annapragada, A.; Vigneswaran, N. Cancer stem cells are enriched in Fanconi anemia head and neck squamous cell carcinomas. Int. J. Oncol. 2014, 45, 2365–2372.
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33.
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9.
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452.
- Chen, Y.; Li, D.; Wang, D.; Liu, X.; Yin, N.; Song, Y.; Lu, S.H.; Ju, Z.; Zhan, Q. Quiescence and attenuated DNA damage response promote survival of esophageal cancer stem cells. J. Cell Biochem. 2012, 113, 3643–3652.
- Maugeri-Sacca, M.; Bartucci, M.; De Maria, R. DNA damage repair pathways in cancer stem cells. Mol. Cancer Ther. 2012, 11, 1627–1636.
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37.
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659.
- Atun, R.; Jaffray, D.A.; Barton, M.B.; Bray, F.; Baumann, M.; Vikram, B.; Hanna, T.P.; Knaul, F.M.; Lievens, Y.; Lui, T.Y.; et al. Expanding global access to radiotherapy. Lancet Oncol. 2015, 16, 1153–1186.
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964.
- Sorolla, M.A.; Parisi, E.; Sorolla, A. Determinants of Sensitivity to Radiotherapy in Endometrial Cancer. Cancers 2020, 12, 1906.
- Seshacharyulu, P.; Baine, M.J.; Souchek, J.J.; Menning, M.; Kaur, S.; Yan, Y.; Ouellette, M.M.; Jain, M.; Lin, C.; Batra, S.K. Biological determinants of radioresistance and their remediation in pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 69–92.
- MacLachlan, T.K.; Takimoto, R.; El-Deiry, W.S. BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol. Cell. Biol. 2002, 22, 4280–4292.
- Wang, Y.Y.; Chen, Y.K.; Lo, S.; Chi, T.C.; Chen, Y.H.; Hu, S.C.; Chen, Y.W.; Jiang, S.S.; Tsai, F.Y.; Liu, W.; et al. MRE11 promotes oral cancer progression through RUNX2/CXCR4/AKT/FOXA2 signaling in a nuclease-independent manner. Oncogene 2021, 40, 3510–3532.
- Kamarli, A.P.; Davliatkadamov, S. Hypodermin-chlorophos against warble fly larvae in yaks. Veterinariia 1975, 10, 64–65.
- Chen, Y.C.; Su, Y.N.; Chou, P.C.; Chiang, W.C.; Chang, M.C.; Wang, L.S.; Teng, S.C.; Wu, K.J. Overexpression of NBS1 contributes to transformation through the activation of phosphatidylinositol 3-kinase/Akt. J. Biol. Chem. 2005, 280, 32505–32511.
- Yang, M.H.; Chang, S.Y.; Chiou, S.H.; Liu, C.J.; Chi, C.W.; Chen, P.M.; Teng, S.C.; Wu, K.J. Overexpression of NBS1 induces epithelial-mesenchymal transition and co-expression of NBS1 and Snail predicts metastasis of head and neck cancer. Oncogene 2007, 26, 1459–1467.
- Ho, S.S.; Zhang, W.Y.; Tan, N.Y.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189.
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072.
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518.
- Wan, D.; Jiang, W.; Hao, J. Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front. Immunol. 2020, 11, 615.
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414.
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mule, J.J.; Barber, G.N. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018, 37, 2037–2051.
- Suter, M.A.; Tan, N.Y.; Thiam, C.H.; Khatoo, M.; MacAry, P.A.; Angeli, V.; Gasser, S.; Zhang, Y.L. cGAS-STING cytosolic DNA sensing pathway is suppressed by JAK2-STAT3 in tumor cells. Sci. Rep. 2021, 11, 7243.
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell Mol. Biol. Lett. 2018, 23, 12.
- Hu, B.; Jin, C.; Li, H.B.; Tong, J.; Ouyang, X.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768.
- Fernandes-Alnemri, T.; Yu, J.W.; Juliana, C.; Solorzano, L.; Kang, S.; Wu, J.; Datta, P.; McCormick, M.; Huang, L.; McDermott, E.; et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010, 11, 385–393.
- Karpova, A.Y.; Trost, M.; Murray, J.M.; Cantley, L.C.; Howley, P.M. Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc. Natl. Acad. Sci. USA 2002, 99, 2818–2823.
- Huang, T.T.; Wuerzberger-Davis, S.M.; Wu, Z.H.; Miyamoto, S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell 2003, 115, 565–576.
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386.
- Wolf, C.; Rapp, A.; Berndt, N.; Staroske, W.; Schuster, M.; Dobrick-Mattheuer, M.; Kretschmer, S.; Konig, N.; Kurth, T.; Wieczorek, D.; et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat. Commun. 2016, 7, 11752.
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017, 45, 4590–4605.
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522.
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139.
- Reislander, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143.
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618.
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109.
- Gemenetzidis, E.; Gammon, L.; Biddle, A.; Emich, H.; Mackenzie, I.C. Invasive oral cancer stem cells display resistance to ionising radiation. Oncotarget 2015, 6, 43964–43977.
- Zhang, M.; Behbod, F.; Atkinson, R.L.; Landis, M.D.; Kittrell, F.; Edwards, D.; Medina, D.; Tsimelzon, A.; Hilsenbeck, S.; Green, J.E.; et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008, 68, 4674–4682.
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. JCI Insight 2018, 3.
- Abad, E.; Civit, L.; Potesil, D.; Zdrahal, Z.; Lyakhovich, A. Enhanced DNA damage response through RAD50 in triple negative breast cancer resistant and cancer stem-like cells contributes to chemoresistance. FEBS J. 2021, 288, 2184–2202.
- Borgmann, K.; Kocher, S.; Kriegs, M.; Mansour, W.Y.; Parplys, A.C.; Rieckmann, T.; Rothkamm, K. DNA Repair. Recent. Results Cancer Res. 2016, 198, 1–24.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760.
- Valencia-Gonzalez, H.A.; Ruiz, G.; Ortiz-Sanchez, E.; Garcia-Carranca, A. Cancer Stem Cells from Tumor Cell Lines Activate the DNA Damage Response Pathway after Ionizing Radiation More Efficiently Than Noncancer Stem Cells. Stem Cells Int. 2019, 2019, 7038953.
- Kim, H.; Lin, Q.; Yun, Z. BRCA1 regulates the cancer stem cell fate of breast cancer cells in the context of hypoxia and histone deacetylase inhibitors. Sci. Rep. 2019, 9, 9702.
- Gorodetska, I.; Lukiyanchuk, V.; Peitzsch, C.; Kozeretska, I.; Dubrovska, A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int. J. Cancer 2019, 145, 2974–2985.
- Anuja, K.; Chowdhury, A.R.; Saha, A.; Roy, S.; Rath, A.K.; Kar, M.; Banerjee, B. Radiation-induced DNA damage response and resistance in colorectal cancer stem-like cells. Int. J. Radiat. Biol. 2019, 95, 667–679.
- Nathansen, J.; Lukiyanchuk, V.; Hein, L.; Stolte, M.I.; Borgmann, K.; Lock, S.; Kurth, I.; Baumann, M.; Krause, M.; Linge, A.; et al. Oct4 confers stemness and radioresistance to head and neck squamous cell carcinoma by regulating the homologous recombination factors PSMC3IP and RAD54L. Oncogene 2021, 40, 4214–4228.
- Cheng, L.; Wu, Q.; Huang, Z.; Guryanova, O.A.; Huang, Q.; Shou, W.; Rich, J.N.; Bao, S. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. EMBO J. 2011, 30, 800–813.
- Lou, D.; Zhu, L.; Ding, H.; Dai, H.Y.; Zou, G.M. Aberrant expression of redox protein Ape1 in colon cancer stem cells. Oncol. Lett. 2014, 7, 1078–1082.
- Pedersen, H.; Anne Adanma Obara, E.; Elbaek, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. Int. J. Mol. Sci. 2020, 21, 1388.
- Xu, X.L.; Xing, B.C.; Han, H.B.; Zhao, W.; Hu, M.H.; Xu, Z.L.; Li, J.Y.; Xie, Y.; Gu, J.; Wang, Y.; et al. The properties of tumor-initiating cells from a hepatocellular carcinoma patient’s primary and recurrent tumor. Carcinogenesis 2010, 31, 167–174.
- Tse, A.N.; Carvajal, R.; Schwartz, G.K. Targeting checkpoint kinase 1 in cancer therapeutics. Clin. Cancer Res. 2007, 13, 1955–1960.
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511.
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190.
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979.
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100.
- Sun, L.L.; Yang, R.Y.; Li, C.W.; Chen, M.K.; Shao, B.; Hsu, J.M.; Chan, L.C.; Yang, Y.; Hsu, J.L.; Lai, Y.J.; et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 2018, 8, 1307–1316.
- Tang, Z.; Pilie, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909.
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751.
- Zhou, C.; Lin, A.; Cao, M.; Ding, W.; Mou, W.; Guo, N.; Chen, Z.; Zhang, J.; Luo, P. Activation of the DDR Pathway Leads to the Down-Regulation of the TGFbeta Pathway and a Better Response to ICIs in Patients With Metastatic Urothelial Carcinoma. Front. Immunol. 2021, 12, 634741.
- Song, Y.; Huang, J.; Liang, D.; Hu, Y.; Mao, B.; Li, Q.; Sun, H.; Yang, Y.; Zhang, J.; Zhang, H.; et al. DNA Damage Repair Gene Mutations Are Indicative of a Favorable Prognosis in Colorectal Cancer Treated With Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 549777.
- Muller, L.; Tunger, A.; Plesca, I.; Wehner, R.; Temme, A.; Westphal, D.; Meier, F.; Bachmann, M.; Schmitz, M. Bidirectional Crosstalk Between Cancer Stem Cells and Immune Cell Subsets. Front. Immunol. 2020, 11, 140.
- Wei, Q.; Frazier, M.L.; Levin, B. DNA repair: A double-edged sword. J. Natl. Cancer Inst. 2000, 92, 440–441.
- Hutchinson, M.N.D.; Mierzwa, M.; D’Silva, N.J. Radiation resistance in head and neck squamous cell carcinoma: Dire need for an appropriate sensitizer. Oncogene 2020, 39, 3638–3649.
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104.
- Huang, J. Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol. Ther. 2021, 220, 107720.
- Kang, J.; D’Andrea, A.D.; Kozono, D. A DNA repair pathway-focused score for prediction of outcomes in ovarian cancer treated with platinum-based chemotherapy. J. Natl. Cancer Inst. 2012, 104, 670–681.
- Pitroda, S.P.; Pashtan, I.M.; Logan, H.L.; Budke, B.; Darga, T.E.; Weichselbaum, R.R.; Connell, P.P. DNA repair pathway gene expression score correlates with repair proficiency and tumor sensitivity to chemotherapy. Sci. Transl. Med. 2014, 6, 229ra242.
- Bold, I.T.; Specht, A.K.; Droste, C.F.; Zielinski, A.; Meyer, F.; Clauditz, T.S.; Munscher, A.; Werner, S.; Rothkamm, K.; Petersen, C.; et al. DNA Damage Response during Replication Correlates with CIN70 Score and Determines Survival in HNSCC Patients. Cancers 2021, 13, 1194.


