 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Amalendu Ghosh | + 3780 word(s) | 3780 | 2021-10-11 04:25:23 | | | |
| 2 | Vivi Li | Meta information modification | 3780 | 2021-10-15 04:57:45 | | |
Video Upload Options
Thrips are insect pests of economically important agricultural, horticultural, and forest crops. They cause damage by sucking plant sap and by transmitting several tospoviruses, ilarviruses, carmoviruses, sobemoviruses, and machlomoviruses. Accurate and timely identification is the key to successful management of thrips species. However, their small size, cryptic nature, presence of color and reproductive morphs, and intraspecies genetic variability make the identification of thrips species challenging. The use of molecular and electronic detection platforms has made thrips identification rapid, precise, sensitive, high throughput, and independent of developmental stages. Multi-locus phylogeny based on mitochondrial, nuclear, and other markers has resolved ambiguities in morphologically indistinguishable thrips species. Microsatellite, RFLP, RAPD, AFLP, and CAPS markers have helped to explain population structure, gene flow, and intraspecies heterogeneity. Recent techniques such as LAMP and RPA have been employed for sensitive and on-site identification of thrips. Artificial neural networks and high throughput diagnostics facilitate automated identification.
1. Introduction
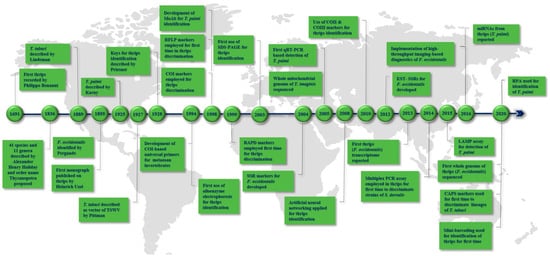
2. Landmarks in Thrips Diagnostics
3. PCR-Based Identification of Thrips Using Molecular Markers
3.1. COI Markers
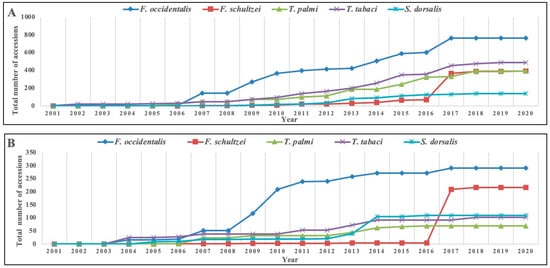
3.2. Thrips Genetic Diversity Studies Using COI Markers
3.2.1. T. tabaci
3.2.2. T. palmi
3.3. COII Markers
3.4. COIII Markers
3.5. rRNA-ITS
3.6. Other Marker Genes Used for Thrips Identification
3.7. SSR/Microsatellite Markers
3.8. RFLP Markers
3.9. RAPD Markers
3.10. AFLP Markers
References
- Mound, L.A. Thysanoptera (Thrips) World Checklist. Available online: https://www.ento.csiro.au/thysanoptera/worldthrips.php (accessed on 20 July 2021).
- Mound, L.A.; Morris, D.C. The insect order Thysanoptera: Classification versus systematics. Zootaxa 2007, 1668, 395–411.
- Nel, P.; Peñalver, E.; Azar, D.; Hodebert, G.; Nel, A. Modern thrips families Thripidae and Phlaeothripidae in early Cretaceous amber (Insecta: Thysanoptera). Ann. Soc. Entomol. Fr. 2013, 46, 154–163.
- Cook, D.; Herbert, A.; Akin, D.S.; Reed, J. Biology, crop injury, and management of thrips (Thysanoptera: Thripidae) infesting cotton seedlings in the United States. J. Integr. Pest Manag. 2011, 2, B1–B9.
- Sampson, C.; Kirk, W.D.J. Predatory mites double the economic injury level of Frankliniella occidentalis in strawberry. BioControl 2016, 61, 661–669.
- Parrella, G.; Gognalons, P.; Gebre-Selassiè, K.; Vovlas, C.; Marchoux, G. An update of the host range of tomato spotted wilt virus. J. Plant Pathol. 2003, 85, 227–264.
- Pappu, H.R.; Jones, R.A.C.; Jain, R.K. Global status of tospovirus epidemics in diverse cropping systems: Successes achieved and challenges ahead. Virus Res. 2009, 141, 219–236.
- Oliver, J.E.; Whitfield, A.E. The genus tospovirus: Emerging Bunyaviruses that threaten food security. Annu. Rev. Virol. 2016, 3, 101–124.
- Sherwood, J.L.; German, T.L.; Moyer, J.W.; Ullman, D.E. Tomato spotted wilt. Plant Health Instr. 2003.
- Riley, D.G.; Joseph, S.V.; Srinivasan, R.; Diffie, S. Thrips vectors of tospoviruses. J. Integr. Pest Manag. 2011, 2, I1–I10.
- Adkins, S. Tomato spotted wilt virus—Positive steps towards negative success. Mol. Plant Pathol. 2000, 1, 151–157.
- Reddy, D.V.R.; Buiel, A.A.M.; Satyanarayana, T.; Dwivedi, S.L.; Reddy, A.S.; Ratna, A.S.; Vijayalakshmi, K.; Ranga Rao, G.V.; Naidu, R.A.; Wightman, J.A. Peanut bud necrosis disease: An overview. In Recent Studies on Peanut Bud Necrosis Disease; ICRISAT Asia Centre: Patancheru, Andhra Pradesh, India, 1995; pp. 3–7.
- Brunner, P.C.; Chatzivassiliou, E.K.; Katis, N.I.; Frey, J.E. Host-associated genetic differentiation in Thrips tabaci (Insecta; Thysanoptera), as determined from mtDNA sequence data. Heredity 2004, 93, 364–370.
- Hoddle, M.S.; Heraty, J.M.; Rugman-Jones, P.F.; Mound, L.A.; Stouthamer, R. Relationships among species of Scirtothrips (Thysanoptera: Thripidae, Thripinae) using molecular and morphological data. Ann. Entomol. Soc. Am. 2008, 101, 491–500.
- Brunner, P.C.; Frey, J.E. Habitat-specific population structure in native western flower thrips Frankliniella occidentalis (Insecta, Thysanoptera). J. Evol. Biol. 2010, 23, 797–804.
- Rugman-Jones, P.F.; Hoddle, M.S.; Stouthamer, R. Nuclear-mitochondrial barcoding exposes the global pest western flower thrips (Thysanoptera: Thripidae) as two sympatric cryptic species in its native California. J. Econ. Entomol. 2010, 103, 877–886.
- Jacobson, A.L.; Booth, W.; Vargo, E.L.; Kennedy, G.G. Thrips tabaci population genetic structure and polyploidy in relation to competency as a vector of tomato spotted wilt virus. PLoS ONE 2013, 8, e54484.
- Moritz, G. Pictorial key to the economically important species of Thysanoptera in Central Europe. EPPO Bull. 1994, 24, 181–208.
- Mound, L.A.; Kibby, G. Thysanoptera: An Identification Guide; CABI International: Wallingford, UK, 1998.
- Bhatti, J.S. Yellow dorsally spotted species of thrips (Terebrantia: Thripidae) in India with description of a new species in flowers of Tabernaemontana (Apocynaceae) and Lantana (Verbenaceae). Thrips 1999, 1, 58–65.
- Mound, L.A.; Masumoto, M. The genus Thrips (Thysanoptera, Thripidae) in Australia, New Caledonia and New Zealand. Zootaxa 2005, 1020, 1–64.
- Chandra, M.; Verma, R.K. Key for identification of adult female of Scirtothrips dorsalis (Thysanoptera: Thripidae) based on external morphology. World Appl. Sci. J. 2010, 9, 25.
- Moritz, G.; Subramanian, S.; Brandt, S.; Triapitsyn, S.V. Development of a user-friendly identification system for the native and invasive pest thrips and their parasitoids in east Africa. Phytopathology 2011, 101, 59–60.
- Zhang, H.; Xie, Y.; Li, Z. Identification key to species of Thrips genus from China (Thysanoptera, Thripidae), with seven new records. Zootaxa 2011, 2810, 37–46.
- Skarlinsky, T.; Funderburk, J. A key to some Frankliniella (Thysanoptera: Thripidae) larvae found in Florida with descriptions of the first instar of select species. Fla. Entomol. 2016, 99, 463–470.
- Mound, L.; Nakahara, S.; Tsuda, D.M. Thysanoptera-Terebrantia of the Hawaiian Islands: An identification manual. Zookeys 2016, 549, 71.
- Cluever, J.D.; Smith, H.A. A photo-based key of thrips (Thysanoptera) associated with horticultural crops in Florida. Fla. Entomol. 2017, 100, 454–467.
- Belaam-Kort, I.; Marullo, R.; Attia, S.; Boulahia-Kheder, S. Thrips fauna in citrus orchard in Tunisia: An up-to-date. Bull. Insectology 2020, 73, 1–10.
- Banks, J.N.; Collins, D.W.; Rizvi, R.H.; Northway, B.J.; Danks, C. Production and characterization of monoclonal antibodies against the EU-listed pest Thrips palmi. Food Agric. Immunol. 1998, 10, 281–290.
- Moritz, G.; Delker, C.; Paulsen, M.; Mound, L.A.; Burgermeister, W. Modern methods for identification of Thysanoptera. EPPO Bull. 2000, 30, 591–593.
- Bayar, K.; Törjék, O.; Kiss, E.; Gyulai, G.; Heszky, L. Intra- and interspecific molecular polymorphism of thrips species. Acta Biol. Hung. 2002, 53, 317–324.
- Brunner, P.C.; Fleming, C.; Frey, J.E. A molecular identification key for economically important thrips species (Thysanoptera: Thripidae) using direct sequencing and a PCR-RFLP-based approach. Agric. For. Entomol. 2002, 4, 127–136.
- Toda, S.; Komazaki, S. Identification of thrips species (Thysanoptera: Thripidae) on Japanese fruit trees by polymerase chain reaction and restriction fragment length polymorphism of the ribosomal ITS2 region. Bull. Entomol. Res. 2002, 92, 359–363.
- Walsh, K.; Boonham, N.; Barker, I.; Collins, D.W. Development of a sequence-specific real-time PCR to the melon thrips Thrips palmi (Thysan., Thripidae). J. Appl. Entomol. 2005, 129, 272–279.
- Rugman-Jones, P.F.; Hoddle, M.S.; Mound, L.A.; Stouthamer, R. Molecular identification key for pest species of Scirtothrips (Thysanoptera: Thripidae). J. Econ. Entomol. 2006, 99, 1813–1819.
- XiangQin, M.; LiAng, M.; FangHao, W.; ZhongShi, Z.; WenKai, W.; GuiFen, W. SCAR marker for rapid identification of the western flower thrips, Frankliniella occidentalis (Pergande) (Thysanoptera: Thripidae). Acta Entomol. Sin. 2010, 53, 323–330.
- Przybylska, A.; Fiedler, Ż.; Kucharczyk, H.; Obrępalska-Stęplowska, A. Detection of the quarantine species Thrips palmi by loop-mediated isothermal amplification. PLoS ONE 2015, 10, e0122033.
- Mehle, N.; Trdan, S. Traditional and modern methods for the identification of thrips (Thysanoptera) species. J. Pest Sci. 2012, 85, 179–190.
- Uzel, J. Monografie řádu Thysanoptera; Selbstverlag des Verfassers, Králové: B. E. Tolmana, Hradec Králové, Czech Republic, 1895.
- Pittman, H.A. Spotted wilt of tomatoes. J. Aust. Counc. Sci. Ind. Res. 1927, 1, 74–77.
- Lewis, T.; Loxdale, H.D.; Brookes, C.P. Studying dispersal and persistence of pear thrips populations using genetic markers (allozymes). Courier Forschungsinstitut Senckenberg 1994, 178, 75–78.
- Crespi, B.J.; Carmean, D.A.; Mound, L.A.; Worobey, M.; Morris, D. Phylogenetics of social behavior in Australian gall-forming thrips: Evidence from mitochondrial DNA sequence, adult morphology and behavior, and gall morphology. Mol. Phylogenet. Evol. 1998, 9, 163–180.
- Fedor, P.; Malenovský, I.; Vaňhara, J.; Sierka, W.; Havel, J. Thrips (Thysanoptera) identification using artificial neural networks. Bull. Entomol. Res. 2008, 98, 437–447.
- Asokan, R.; Krishna Kumar, N.K.; Kumar, V.; Ranganath, H.R. Molecular differences in the mitochondrial cytochrome oxidase I (mtCOI) gene and development of a species-specific marker for onion thrips, Thrips tabaci Lindeman, and melon thrips, T. palmi Karny (Thysanoptera: Thripidae), vectors of tospoviruses (Bunyaviridae). Bull. Entomol. Res. 2007, 97, 461–470.
- Inoue, T.; Sakurai, T. The phylogeny of thrips (Thysanoptera: Thripidae) based on partial sequences of cytochrome oxidase I, 28S ribosomal DNA and elongation factor-1 α and the association with vector competence of tospoviruses. Appl. Entomol. Zool. 2007, 42, 71–81.
- Agamy, E.; El-Husseini, M.; El- Sebaey, I.; Wafy, M. Molecular identification of thripids attacking olive groves at Ismailia, Egypt. Egypt. Acad. J. Biol. Sci. A Entomol. 2017, 10, 43–55.
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B Biol. Sci. 2003, 270, 313–321.
- Hebert, P.D.N.; Ratnasingham, S.; DeWaard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. B Biol. Sci. 2003, 270, S96–S99.
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299.
- Timm, A.E.; Stiller, M.; Frey, J.E. A molecular identification key for economically important thrips species (Thysanoptera: Thripidae) in southern Africa. Afr. Entomol. 2008, 16, 68–75.
- Chakraborty, R.; Singha, D.; Kumar, V.; Pakrashi, A.; Kundu, S.; Chandra, K.; Patnaik, S.; Tyagi, K. DNA barcoding of selected Scirtothrips species (Thysanoptera) from India. Mitochondrial DNA Part B 2019, 4, 2710–2714.
- Cubillos-Salamanca, Y.P.; Rodríguez-Maciel, J.C.; Pineda-Guillermo, S.; Silva-Rojas, H.V.; Berzosa, J.; Tejeda-Reyes, M.A.; Rebollar-Alviter, Á. Identification of thrips species and resistance of Frankliniella occidentalis (Thysanoptera: Thripidae) to malathion, spinosad, and bifenthrin in blackberry crops. Fla. Entomol. 2019, 102, 738–746.
- Bravo-Pérez, D.; Santillán-Galicia, M.T.; Johansen-Naime, R.M.; González-Hernández, H.; Segura-León, O.L.; Ochoa-Martínez, D.L.; Guzman-Valencia, S. Species diversity of thrips (Thysanoptera) in selected avocado orchards from Mexico based on morphology and molecular data. J. Integr. Agric. 2018, 17, 2509–2517.
- Karimi, J.; Hassani-Kakhki, M.; Modarres Awal, M. Identifying thrips (Insecta: Thysanoptera) using DNA barcodes. J. Cell Mol. Res. 2010, 2, 35–41.
- Zhang, G.F.; Meng, X.Q.; Min, L.; Qiao, W.N.; Wan, F.H. Rapid diagnosis of the invasive species, Frankliniella occidentalis (Pergande): A species-specific COI marker. J. Appl. Entomol. 2012, 136, 410–420.
- Leão, E.U.; de Almeida Spadotti, D.M.; Rocha, K.C.G.; Lima, E.F.B.; Tavella, L.; Turina, M.; Krause-Sakate, R. Efficient detection of Frankliniella schultzei (Thysanoptera, Thripidae) by cytochrome oxidase I gene (mtCOI) direct sequencing and real-time PCR. Braz. Arch. Biol. Technol. 2017, 60, e17160425.
- Xie, Y.; Mound, L.A.; Zhang, H. A new species of Heliothrips (Thysanoptera, Panchaetothripinae), based on morphological and molecular data. Zootaxa 2019, 4638, 143–150.
- Tyagi, K.; Kumar, V.; Singha, D.; Chakraborty, R.; Muehlethaler, R. Morphological and DNA barcoding evidence for invasive pest thrips, Thrips parvispinus (Thripidae: Thysanoptera), newly recorded from India. J. Insect Sci. 2015, 15, 105.
- Suganthy, M.; Rageshwari, S.; Senthilraja, C.; Nakkeeran, S.; Malathi, V.G.; Ramaraju, K.; Renukadevi, P. New record of western flower thrips, Frankliniella occidentalis (Pergande) (Thysanoptera: Thripidae) in south India. Int. J. Environ. Agric. Biotechnol. 2016, 1, 857–867.
- Singha, D.; Tyagi, K.; Kumar, V. First record of Podothrips erami (Thysanoptera: Tubulifera) from India. Halteres 2017, 8, 30–32.
- Kobayashi, K.; Hasegawa, E. Discrimination of reproductive forms of Thrips tabaci (Thysanoptera: Thripidae) by PCR with sequence specific primers. J. Econ. Entomol. 2012, 105, 555–559.
- Kumar, V.; Seal, D.R.; Osborne, L.S.; McKenzie, C.L. Coupling scanning electron microscopy with DNA bar coding: A novel approach for thrips identification. Appl. Entomol. Zool. 2014, 49, 403–409.
- Zhang, D.X.; Hewitt, G.M. Nuclear integrations: Challenges for mitochondrial DNA markers. Trends Ecol. Evol. 1996, 11, 247–251.
- Parfait, B.; Rustin, P.; Munnich, A.; Rötig, A. Coamplification of nuclear pseudogenes and assessment of heteroplasmy of mitochondrial DNA mutations. Biochem. Biophys. Res. Commun. 1998, 247, 57–59.
- Bensasson, D.; Zhang, D.X.; Hartl, D.L.; Hewitt, G.M. Mitochondrial pseudogenes: Evolution’s misplaced witnesses. Trends Ecol. Evol. 2001, 16, 314–321.
- Frey, J.E.; Frey, B. Origin of intra-individual variation in PCR-amplified mitochondrial cytochrome oxidade I of Thrips tabaci (Thysanoptera: Thripidae): Mitochondrial heteroplasmy or nuclear integration? Hereditas 2004, 140, 92–98.
- Marullo, R.; Mercati, F.; Vono, G. DNA barcoding: A reliable method for the identification of thrips species (Thysanoptera, Thripidae) collected on sticky traps in onion fields. Insects 2020, 11, 489.
- Jenser, G.; Szénási, Á. Review of the biology and vector capability of Thrips tabaci Lindeman (Thysanoptera: Thripidae). Acta Phytopathol. Entomol. Hung. 2004, 39, 137–155.
- Murai, T. Parthenogenetic reproduction in Thrips tabaci and Frankliniella intonsa (Insecta: Thysanoptera). In Advances in Invertebrate Reproduction 5; Hoshi, M., Yamashita, O., Eds.; Elsevier: Amsterdam, The Netherlands, 1990; pp. 357–362.
- Zawirska, I. Untersuchungen über zwei biologische Typen von Thrips tabaci Lind. (Thysanoptera, Thripidae) in der VR Polen. Arch. Phytopathol. Plant Prot. 1976, 12, 411–422.
- Chatzivassiliou, E.K. Thrips tabaci: An ambiguous vector of TSWV in perspective. In Thrips and Tospoviruses: Proceedings of the 7th International Symposium on Thysanoptera, Reggio Calabria, Italy, 2–7 July 2001; Marullo, R., Mound, L., Eds.; ANIC: Canberra, Australia, 2002; pp. 69–75.
- Srinivasan, R.; Guo, F.; Riley, D.; Diffie, S.; Gitaitis, R.; Sparks, A.; Jeyaprakash, A. Assessment of variation among Thrips tabaci populations from Georgia and Peru based on polymorphism in mitochondrial cytochrome oxidase I and ribosomal ITS2 sequences. J. Entomol. Sci. 2011, 46, 191–203.
- Fekrat, L.; Manzari, S.; Shishehbor, P. Morphometric and molecular variation in Thrips tabaci Lindeman (Thysanoptera: Thripidae) populations on onion and tobacco in Iran. J. Agric. Sci. Technol. 2014, 16, 1505–1516.
- Li, X.; Zhang, Z.; Zhang, J.; Huang, J.; Wang, L.; Li, Y.; Hafeez, M.; Lu, Y. Population genetic diversity and structure of Thrips tabaci (Thysanoptera: Thripidae) on allium hosts in China, inferred from mitochondrial COI gene sequences. J. Econ. Entomol. 2020, 113, 1426–1435.
- Toda, S.; Murai, T. Phylogenetic analysis based on mitochondrial COI gene sequences in Thrips tabaci Lindeman (Thysanoptera: Thripidae) in relation to reproductive forms and geographic distribution. Appl. Entomol. Zool. 2007, 42, 309–316.
- Sogo, K.; Miura, K.; Aizawa, M.; Watanabe, T.; Stouthamer, R. Genetic structure in relation to reproduction mode in Thrips tabaci (Insecta: Thysanoptera). Appl. Entomol. Zool. 2015, 50, 73–77.
- Westmore, G.C.; Poke, F.S.; Allen, G.R.; Wilson, C.R. Genetic and host-associated differentiation within Thrips tabaci Lindeman (Thysanoptera: Thripidae) and its links to tomato spotted wilt virus-vector competence. Heredity 2013, 111, 210–215.
- Nault, B.A.; Kain, W.C.; Wang, P. Seasonal changes in Thrips tabaci population structure in two cultivated hosts. PLoS ONE 2014, 9, e101791.
- Jacobson, A.L.; Nault, B.A.; Vargo, E.L.; Kennedy, G.G. Restricted gene flow among lineages of Thrips tabaci supports genetic divergence among cryptic species groups. PLoS ONE 2016, 11, e0163882.
- Grazia, A.D.; Marullo, R.; Frey, J.E. Preliminary results of molecular polymorphism in field populations of Thrips tabaci Lindeman (Thysanoptera: Thripidae), occurring on onion crops in South Italy. Bodenkult. J.L. Manag. Food Environ. 2015, 66, 12–16.
- Nault, B.A.; Shelton, A.M.; Gangloff-kaufmann, J.L.; Clark, M.E.; Werren, J.L.; Cabrera-la Rosa, J.C.; Kennedy, G.G. Reproductive modes in onion thrips (Thysanoptera: Thripidae) populations from New York onion fields. Environ. Entomol. 2006, 35, 1264–1271.
- Glover, R.H.; Collins, D.W.; Walsh, K.; Boonham, N. Assessment of loci for DNA barcoding in the genus thrips (Thysanoptera:Thripidae). Mol. Ecol. Resour. 2010, 10, 51–59.
- Kadirvel, P.; Srinivasan, R.; Hsu, Y.C.; Su, F.C.; Del La Peña, R. Application of cytochrome oxidase I sequences for phylogenetic analysis and identification of thrips species occurring on vegetable crops. J. Econ. Entomol. 2013, 106, 408–418.
- Rebijith, K.B.; Asokan, R.; Krishna, V.; Ranjitha, H.H.; Krishna Kumar, N.K.; Ramamurthy, V.V. DNA barcoding and elucidation of cryptic diversity in thrips (Thysanoptera). Fla. Entomol. 2014, 97, 1328–1347.
- Iftikhar, R.; Ashfaq, M.; Rasool, A.; Hebert, P.D.N. DNA barcode analysis of thrips (Thysanoptera) diversity in Pakistan reveals cryptic species complexes. PLoS ONE 2016, 11, e0146014.
- Ghosh, A.; Jagdale, S.S.; Basavaraj; Dietzgen, R.G.; Jain, R.K. Genetics of Thrips palmi (Thysanoptera: Thripidae). J. Pest Sci. 2020, 93, 27–39.
- Tyagi, K.; Kumar, V.; Singha, D.; Chandra, K.; Laskar, B.A.; Kundu, S.; Chakraborty, R.; Chatterjee, S. DNA Barcoding studies on thrips in India: Cryptic species and species complexes. Sci. Rep. 2017, 7, 4898.
- Gholamzadeh, S.; Incekara, Ü. Review of molecular taxonomy studies on Coleoptera aquatic insects. Int. J. Entomol. Res. 2016, 4, 25–36.
- Zoldos, V.; Papes, D.; Cerbah, M.; Panaud, O.; Besendorfer, V.; Siljak-Yakovlev, S. Molecular-cytogenetic studies of ribosomal genes and heterochromatin reveal conserved genome organization among 11 Quercus species. Theor. Appl. Genet. 1999, 99, 969–977.
- Farris, R.E.; Ruiz-Arce, R.; Ciomperlik, M.; Vasquez, J.D.; DeLeón, R. Development of a ribosomal DNA ITS2 marker for the identification of the thrips, Scirtothrips dorsalis. J. Insect Sci. 2010, 10, 26.
- Jenser, G.; Almási, A.; Tóbiás, I. Host range and number of generations of pea thrips (Kakothrips pisivorus westwood, 1880) (Thysanoptera: Thripidae) in Hungary. Acta Phytopathol. Entomol. Hung. 2012, 47, 97–102.
- Seepiban, C.; Charoenvilaisiri, S.; Kumpoosiri, M.; Bhunchoth, A.; Chatchawankanphanich, O.; Gajanandana, O. Development of a protocol for the identification of tospoviruses and thrips species in individual thrips. J. Virol. Methods 2015, 222, 206–213.
- Kabir, M.T.; Snow, J.W. Phoresy or an accident? Trafficking of flower-feeding thrips by pollen-foraging bees. Ecology 2019, 100, e02671.
- Barba-Alvarado, A.A.; Jaén-Sanjur, J.N.; Galipienso, L.; Elvira-González, L.; Rubio, L.; Herrera-Vásquez, J.A. Molecular identification, occurrence and distribution of Thrips palmi, Frankliniella intonsa and Frankliniella cephalica (Thysanoptera: Thripidae) on cucurbit crops in Panama. J. Plant Prot. Res. 2020, 60, 68–76.
- Kang, T.J.; Ahn, S.J.; An, T.J.; Cho, M.R.; Jeon, H.Y.; Jung, J.A. Thrips in medicinal crops in Korea: Identification and their damages. Korean J. Med. Crop Sci. 2012, 20, 487–492.
- Hu, T.; Rong, Z.; Jinliang, Z.; Yusheng, W.; Fanghao, W.; Guifen, Z. Identification of invasive species Frankliniella occidentalis and native species F. intonsa based on double gene markers. Chin. J. Biol. Control 2017, 612–622.
- Gikonyo, M.W.; Niassy, S.; Moritz, G.B.; Khamis, F.M.; Magiri, E.; Subramanian, S. Resolving the taxonomic status of Frankliniella schultzei (Thysanoptera: Thripidae) colour forms in Kenya—A morphological-, biological-, molecular- and ecological-based approach. Int. J. Trop. Insect Sci. 2017, 37, 57–70.
- Latha, K.R.; Krishna Kumar, N.K.; Mahadeva Swamy, H.M.; Asokan, R.; Ranganath, H.R.; Mahmood, R. Molecular identification and diversity of chilli thrips, Scirtothrips dorsalis Hood (Thysanoptera: Thripidae) employing ITS2 marker. Pest Manag. Hortic. Ecosyst. 2015, 21, 16–26.
- Medina, C.D.R.; Apolinario, J.J.G. Localization of populations of Scirtothrips dorsalis Hood collected from various mango-growing areas in the Philippines. Acta Hort. 2017, 1183, 305–310.
- Tseng, L.; Chang, N.; Tseng, M.; Yeh, W. Genetic variation of Thrips tabaci Lindeman (Thysanopetra: Thripidae) in the Pacific Rim. Formos. Entomol. 2010, 30, 219–223.
- Almási, A.; Tóbiás, I.; Bujdos, L.; Jenser, G. Molecular characterisation of Thrips tabaci Lindeman, 1889 (Thysanoptera: Thripidae) populations in Hungary based on the ITS2 sequences. Acta Zool. Acad. Sci. Hung. 2016, 62, 157–164.
- Morris, D.C.; Mound, L.A. Molecular relationships between populations of South African citrus thrips (Scirtothrips aurantii Faure) in South Africa and Queensland, Australia. Aust. J. Entomol. 2004, 43, 353–358.
- Vogler, A.P.; DeSalle, R. Evolution and phylogenetic information content of the ITS-1 region in the tiger beetle Cicindela dorsalis. Mol. Biol. Evol. 1994, 11, 393–405.
- Fenton, B.; Malloch, G.; Germa, F. A study of variation in rDNA ITS regions shows that two haplotypes coexist within a single aphid genome. Genome 1998, 41, 337–345.
- Leo, N.P.; Barker, S.C. Intragenomic variation in ITS2 rDNA in the louse of humans, Pediculus humanus: ITS2 is not a suitable marker for population studies in this species. Insect Mol. Biol. 2002, 11, 651–657.
- Dentinger, B.T.M.; Didukh, M.Y.; Moncalvo, J.-M. Comparing COI and ITS as DNA barcode markers for mushrooms and allies (Agaricomycotina). PLoS ONE 2011, 6, e25081.
- Li, X.-W.; Wang, P.; Fail, J.; Shelton, A.M. Detection of gene flow from sexual to asexual lineages in Thrips tabaci (Thysanoptera: Thripidae). PLoS ONE 2015, 10, e0138353.
- Van der Kooi, C.J.; Schwander, T. Evolution of asexuality via different mechanisms in grass thrips (Thysanoptera: Aptinothrips). Evolution 2014, 68, 1883–1893.
- Buckman, R.S.; Mound, L.A.; Whiting, M.F. Phylogeny of thrips (Insecta: Thysanoptera) based on five molecular loci. Syst. Entomol. 2013, 38, 123–133.
- Fontcuberta García-Cuenca, A.; Dumas, Z.; Schwander, T. Extreme genetic diversity in asexual grass thrips populations. J. Evol. Biol. 2016, 29, 887–899.
- Gupta, P.K.; Varshney, R.K.; Sharma, P.C.; Ramesh, B. Molecular markers and their applications in wheat breeding. Plant Breed. 1999, 118, 369–390.
- Miah, G.; Rafii, M.Y.; Ismail, M.R.; Puteh, A.B.; Rahim, H.A.; Islam, N.K.; Latif, M.A. A review of microsatellite markers and their applications in rice breeding programs to improve blast disease resistance. Int. J. Mol. Sci. 2013, 14, 22499–22528.
- Brunner, P.C.; Frey, J.E. Isolation and characterization of six polymorphic microsatellite loci in the western flower thrips Frankliniella occidentalis (Insecta, Thysanoptera). Mol. Ecol. Notes 2004, 4, 599–601.
- Lyu, Z.; Zhi, J.; Zhou, Y.; Meng, Z.; Wen, J. Population genetic structure and migration patterns of Dendrothrips minowai (Thysanoptera: Thripidae) in Guizhou, China. Entomol. Sci. 2017, 20, 127–136.
- Cao, L.; Gao, Y.; Gong, Y.; Chen, J.; Chen, M.; Hoffmann, A.; Wei, S. Population analysis reveals genetic structure of an invasive agricultural thrips pest related to invasion of greenhouses and suitable climatic space. Evol. Appl. 2019, 12, 1868–1880.
- Yang, X.M.; Lou, H.; Sun, J.T.; Zhu, Y.M.; Xue, X.F.; Hong, X.Y. Temporal genetic dynamics of an invasive species, Frankliniella occidentalis (Pergande), in an early phase of establishment. Sci. Rep. 2015, 5, 11877.
- Duan, H.-S.; Yu, Y.; Zhang, A.-S.; Guo, D.; Tao, Y.-L.; Chu, D. Sudden widespread distribution of Frankliniella occidentalis (Thysanoptera: Thripidae) in Shandong province, China. Fla. Entomol. 2013, 96, 933–940.
- Hondelmann, P.; Nyasani, J.O.; Subramanian, S.; Meyhöfer, R. Genetic structure and diversity of western flower thrips, Frankliniella occidentalis in a French bean agroecosystem of Kenya. Int. J. Trop. Insect Sci. 2017, 37, 71–78.
- Yang, X.-M.; Sun, J.-T.; Xue, X.-F.; Zhu, W.-C.; Hong, X.-Y.; Yang, X.-M.; Sun, J.-T.; Xue, X.-F.; Zhu, W.-C.; Hong, X.-Y. Development and characterization of 18 novel EST-SSRs from the western flower thrips, Frankliniella occidentalis (Pergande). Int. J. Mol. Sci. 2012, 13, 2863–2876.
- Huisheng, D.; Ansheng, Z.; Chuanzhi, Z.; Yi, Y.; Dong, C. Characterization and molecular marker screening of EST-SSRs and their polymorphism compared with Genomic-SSRs in Frankliniella occidentalis (Thysanoptera:Thripidae). Acta Entomol. Sin. 2012, 55, 634–640.
- Liu, J.; Li, Z.; Chen, X.; Huang, H.; Gui, F. Development of polymorphic EST-SSR markers by sequence alignment in Frankliniella occidentalis (Pergande). J. Asia. Pac. Entomol. 2014, 17, 581–585.
- Rugman-Jones, P.F.; Weeks, A.R.; Hoodle, M.S.; Stouthamer, R. Isolation and characterization of microsatellite loci in the avocado thrips Scirtothrips perseae (Thysanoptera: Thripidae). Mol. Ecol. Notes 2005, 5, 644–646.
- Wu, Y.; Liu, K.; Qiu, H.; Li, F.; Cao, Y. Polymorphic microsatellite markers in Thrips hawaiiensis (Thysanoptera: Thripidae). Appl. Entomol. Zool. 2014, 49, 619–622.
- Cao, L.J.; Li, Z.M.; Wang, Z.H.; Zhu, L.; Gong, Y.J.; Chen, M.; Wei, S.J. Bulk development and stringent selection of microsatellite markers in the western flower thrips Frankliniella occidentalis. Sci. Rep. 2016, 6, 26512.
- Gao, Y.; Gong, Y.; Ma, L.; Cao, L.; Chen, J.; Chen, M.; Wei, S.; Gao, Y.; Gong, Y.; Ma, L.; et al. Genome-wide developed microsatellite markers for the melon thrips Thrips palmi Karny (Thysanoptera: Thripidae). Zool. Syst. 2019, 44, 100–110.
- De Grazia, A.; Marullo, R.; Moritz, G. Molecular diagnosis of native and quarantine pest thrips of southern European citrus orchards. Bull. Insectology 2016, 69, 1–6.
- Przybylska, A.; Fiedler, Ż.; Obrępalska-Stęplowska, A. PCR-RFLP method to distinguish Frankliniella occidentalis, Frankliniella intonsa, Frankliniella pallida and Frankliniella tenuicornis. J. Plant Prot. Res. 2016, 56, 60–66.
- Jung, C.R.; Jeong, D.H.; Park, H.W.; Kim, H.J.; Jeon, K.S.; Yoon, J.B. Molecular identification of thrips in two medicinal crops, Cnidium officinale Makino and Ligusticum chuanxiong Hort. Korean J. Med. Crop Sci. 2019, 27, 17–23.
- Moritz, G.; Paulsen, M.; Delker, M.; Picl, S.; Kumm, S. Identification of thrips using ITS-RFLP analysis. In Thrips and Tospoviruses: Proceedings of the 7th International Symposium on Thysanoptera, Reggio Calabria, Italy, 2–7 July 2001; ANIC: Canberra, Australia, 2002; pp. 365–367.
- Takeuchi, R.; Toda, S. Discrimination of two reproductive forms of Thrips tabaci by PCR-RFLP, and distribution of arrhenotokous T. tabaci in Tottori Prefecture. Jpn. J. Appl. Entomol. Zool. 2011, 55, 254–257.
- Hashim, H.O.; Al-Shuhaib, M.B.S. Exploring the potential and limitations of PCR-RFLP and PCR-SSCP for SNP detection: A review. J. Appl. Biotechnol. Rep. 2019, 6, 137–144.
- Williams, J.G.K.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535.
- Klein, M.; Gafni, R. Morphological and molecular variations in thrips population collected on onion plants in Israel. Folia Entomol. Hung. 1996, 57, 57–59.
- Jenser, G.; Szénási, À.; Törjék, O.; Gyulai, G.; Kiss, E.; Heszky, L.; Fail, J. Molecular polymorphism between population of Thrips tabaci Lindeman (Thysanoptera: Thripidae) propagating on tobacco and onion. Acta Phytopathol. Entomol. Hung. 2001, 36, 365–368.
- Ramakrishna Rao, A. Population Dynamics and Molecular Characterization of Thrips and Their Management in Groundnut (Arachis hypogaea L.). Ph.D. Thesis, Acharya, N.G. Ranga Agricultural University, Rajendranagar, Hyderabad, India, 2013.
- Gyulai, G.; Bayar, K.; Törjék, O.; Kiss, J.; Szabó, Z.; Heszky, L. Molecular polymorphism among populations of Frankliniella intonsa. In Thrips and Tospoviruses: Proceedings of the 7th International Symposium on Thysanopetra, Reggio Calabria, Italy, 2–7 July 2001; Marullo, R., Mound, L.A., Eds.; ANIC: Canberra, Australia, 2002; pp. 373–375.
- Brito, R.O.; Artoni, R.F.; Vicari, M.R.; Nogaroto, V.; Silva, J.C.; Matiello, R.R.; Almeida, M.C. Population structure and genetic diversity analysis in Gynaikothrips uzeli (Zimerman, 1909) (Thysanoptera: Phlaeothripidae) by RAPD markers. Bull. Entomol. Res. 2012, 102, 345–351.
- Mainali, B.P.; Shrestha, S.; Lim, U.T.; Kim, Y. Molecular markers of two sympatric species of the genus Frankliniella (Thysanoptera: Thripidae). J. Asia. Pac. Entomol. 2008, 11, 45–48.
- Meena, R.L.; Ramasubram, T.; Venkatesan, S.; Mohankumar, S. Molecular characterization of tospovirus transmitting thrips populations from India. Am. J. Biochem. Biotechnol. 2005, 1, 167–172.
- Jain, S.K.; Neekhra, B.; Pandey, D.; Jain, K. RAPD marker system in insect study: A review. Indian J. Biotechnol. 2010, 9, 7–12.
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; Van De Lee, T.; Hornes, M.; Friters, A.; Pot, J.; Paleman, J.; Kuiper, M.; et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414.
- Wong, A.; Forbes, M.R.; Smith, M.L. Characterization of AFLP markers in damselflies: Prevalence of codominant markers and implications for population genetic applications. Genome 2000, 44, 677–684.
- Reineke, A.; Schmidt, O.; Zebitz, C.P.W. Differential gene expression in two strains of the endoparasitic wasp Venturia canescens identified by cDNA-amplified fragment length polymorphism analysis. Mol. Ecol. 2003, 12, 3485–3492.
- Fang, J.; Kritzman, A.; Yonash, N.; Gera, A.; Pollak, N.; Lavi, U. Genetic variation of thrips populations assessed by amplified fragment length polymorphism (Thysanoptera: Thripidae). Ann. Entomol. Soc. Am. 2005, 98, 351–358.
- Zhang, Z.J.; Wu, Q.J.; Zhang, Y.J.; Lu, Y.B. Genetic differentiation among various populations of Frankliniella occidentalis (Thysanoptera: Thripidae.) assessed by mtDNA sequence and AFLP. In Proceedings of the IXth International Symposium on Thysanoptera and Tospoviruses. J. Insect Sci. 2010, 10, 57.
- Mirnezhad, M.; Schidlo, N.; Klinkhamer, P.G.L.; Leiss, K.A. Variation in genetics and performance of Dutch western flower thrips populations. J. Econ. Entomol. 2012, 105, 1816–1824.


