 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marica Meroni | + 4714 word(s) | 4714 | 2021-10-12 04:26:22 | | | |
| 2 | Peter Tang | Meta information modification | 4714 | 2021-10-15 04:52:19 | | |
Video Upload Options
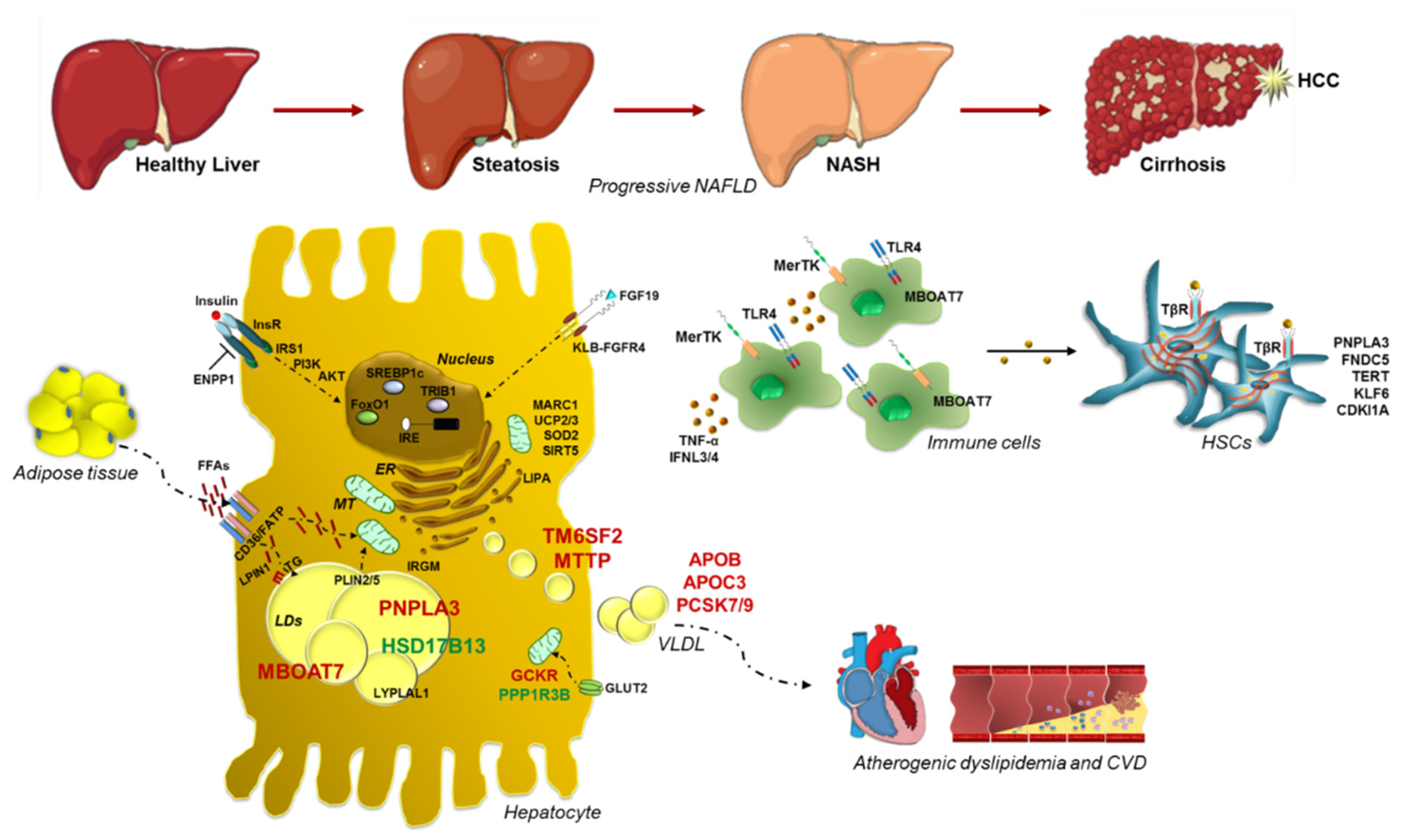
Nonalcoholic fatty liver disease (NAFLD) is the commonest cause of chronic liver disease worldwide. It is closely related to obesity, insulin resistance (IR) and dyslipidemia so much so it is considered the hepatic manifestation of the Metabolic Syndrome. The NAFLD spectrum extends from simple steatosis to nonalcoholic steatohepatitis (NASH), a clinical condition which may progress up to fibrosis, cirrhosis and hepatocellular carcinoma (HCC). NAFLD is a complex disease whose pathogenesis is shaped by both environmental and genetic factors.
1. Introduction
2. Historical Overture to Discover the Link between Genetics and NAFLD
3. Genetic Signature of Glucose and Lipid Metabolism in NAFLD
4. Genetics of Lipid Droplets
5. Advanced Liver Injuries and Genetic Variants
6. Mitochondrial Dysfunctions: The Tipping Point in the Switching from Simple Steatosis to Steatohepatitis
|
Variant |
Gene |
Global MAF |
Function |
Effect |
Impact |
Phenotype |
|---|---|---|---|---|---|---|
|
rs738409 C > G |
PNPLA3 |
0.26 (G) |
Lipid remodeling |
p.I148M |
Loss-of-function |
↑ NAFLD, NASH, fibrosis, HCC |
|
rs58542926 C > T |
TM6SF2 |
0.07 (T) |
VLDL secretion |
p.E167K |
Loss-of-function |
↑ NAFLD, NASH, fibrosis |
|
rs641738 C > T |
TMC4/ MBOAT7 |
0.37 (T) |
Lipid remodeling |
p.G17E |
Loss-of-function |
↑ NAFLD, NASH, fibrosis, HCC |
|
rs1260326 C > T |
GCKR |
0.29 (T) |
Regulation of DNL |
p.P446L |
Loss-of-function |
↑ NAFLD, NASH, fibrosis |
|
rs72613567 T > TA |
HSD17B13 |
0.18 (TA) |
Lipid remodeling |
Truncated protein |
Loss-of-function |
↓ NASH, fibrosis, HCC |
|
rs4841132 G > A |
PPP1R3B |
0.09 (A) |
Glycogen synthesis |
Non-coding |
Gain-of-function |
↓ NAFLD, fibrosis, HCC |
|
rs1801278 C > T |
IRS1 |
0.05 (T) |
Insulin signaling |
p.G972R |
Loss-of-function |
↑ Fibrosis |
|
rs1044498 A > C |
ENPP1 |
0.34 (C) |
Insulin signaling |
p.K121Q |
Gain-of-function |
↑ Fibrosis |
|
rs2954021 G > A |
TRIB1 |
0.45 (A) |
Regulation of DNL |
Non-coding |
Gain-of-function |
↑ NAFLD |
|
rs12137855 C > T |
LYPLAL1 |
0.16 (T) |
Lipid metabolism |
Intronic |
Loss-of-function |
↑ NAFLD |
|
Several |
APOB |
NA |
VLDL secretion |
Protein change |
Loss-of-function |
↑ NAFLD NASH, fibrosis, HCC |
|
Several |
MTTP |
NA |
VLDL secretion |
Protein change |
Loss-of-function |
↑NAFLD |
|
rs236918 G > C |
PCSK7 |
0.26 (C) |
Membrane transferrin receptor shedding and regulation of circulating lipids |
Intronic |
Gain-of-function |
↑ NASH, fibrosis |
|
Several |
PCSK9 |
NA |
LDL uptake |
Protein change |
Loss-of-function |
No evidence of association with steatosis |
|
Several |
LIPA |
NA |
Lipid remodeling |
Protein change |
LAL deficiency |
↑ NAFLD, NASH, fibrosis |
|
rs56225452 G > A |
FATP5 |
0.16 (A) |
FFAs uptake |
Non-coding |
Gain-of-function |
↑ NASH, fibrosis |
|
rs13412852 C > T |
LPIN1 |
0.21 (T) |
Lipid metabolism |
Intronic |
Not Defined |
↓ NASH, fibrosis |
|
rs35568725 A > G |
PLIN2 |
0.02 (G) |
Lipid remodeling |
p.S251P |
Loss-of-function |
↑ NAFLD, NASH, IR, atherosclerosis |
|
rs884164 A > G |
PLIN5 |
0.19 (G) |
Lipid remodeling |
Non-coding |
Loss-of-function |
↑ oxidative stress |
|
rs17618244 G > A |
KLB |
0.15 (A) |
FGF19/FGFR4 pathway |
p.R728Q |
Loss-of-function |
↓ NASH, fibrosis |
|
rs4374383 G > A |
MERTK |
0.45 (A) |
Innate immunity |
Intronic |
Loss-of-function |
↓ Fibrosis |
|
rs3750861 G > A |
KLF6 |
0.07 (A) |
HSCs activation |
Splice variant IVS1-27G |
Loss-of-function |
↓ Fibrosis |
|
Several |
TERT |
NA |
Telomere maintenance |
Protein change |
Loss-of-function |
↑ Fibrosis, HCC |
|
rs12979860 C > T |
IL28B |
0.36 (T) |
Innate immunity |
Alternative IFNL3/4 transcription |
Loss-of-function |
↓ NASH, Fibrosis |
|
rs3480 A > G |
FNDC5 |
0.42 (G) |
HSCs activation |
Non-coding |
Loss-of-function |
↓ Fibrosis |
|
rs4880 C > T |
SOD2 |
0.33 (T) |
Mitochondrial antioxidant |
p.A16V |
Loss-of-function |
↑ Fibrosis |
|
rs695366 G > A |
UCP2 |
0.26 (A) |
Mitochondrial lipid metabolism Oxphos |
−866 promoter variant |
Gain-of-function |
↓ NASH, fibrosis |
|
rs2642438 G > A |
MARC1 |
0.19 (A) |
Mitochondrial detoxification |
p.A165T |
Loss-of-function |
↓ NAFLD, NASH, fibrosis |
MAF: minor allele frequency.
References
- Zobair M. Younossi; Aaron Koenig; Dinan Abdelatif; Yousef Fazel; Linda Henry; Mark Wymer; Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2015, 64, 73-84, 10.1002/hep.28431.
- Mohammed Eslam; Arun J. Sanyal; Jacob George; Brent Neuschwander-Tetri; Claudio Tiribelli; David E. Kleiner; Elizabeth Brunt; Elisabetta Bugianesi; Hannele Yki-Järvinen; Henning Grønbæk; et al.Helena Cortez-PintoJiangao FanLuca ValentiManal AbdelmalekManuel Romero-GomezMary RinellaMarco ArresePierre BedossaPhilip NewsomeQuentin M. AnsteeRajiv JalanRamon BatallerRohit LoombaSilvia SookoianShiv K. SarinStephen HarrisonTakumi KawaguchiVincent Wai-Sun WongVlad RatziuYusuf YilmazZobair Younossi MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999-2014.e1, 10.1053/j.ghttps://doi.org/10.1053/j.gastro.2019.11.312astro.2019.11.312.
- Mohammed Eslam; Philip N. Newsome; Shiv K. Sarin; Quentin M. Anstee; Giovanni Targher; Manuel Romero-Gomez; Shira Zelber-Sagi; Vincent Wai-Sun Wong; Jean-François Dufour; Jörn M. Schattenberg; et al.Takumi KawaguchiMarco ArreseLuca ValentiGamal ShihaClaudio TiribelliHannele Yki-JärvinenJian-Gao FanHenning GrønbækYusuf YilmazHelena Cortez-PintoClaudia P. OliveiraPierre BedossaLeon A. AdamsMing-Hua ZhengYasser FouadWah-Kheong ChanNahum Mendez-SanchezSang Hoon AhnLaurent CasteraElisabetta BugianesiVlad RatziuJacob George A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. Journal of Hepatology 2020, 73, 202-209, 10.1016/j.jhep.2020.03.039.
- Zobair M. Younossi; Linda Henry; Haley Bush; Alita Mishra; Clinical and Economic Burden of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Clinics in Liver Disease 2017, 22, 1-10, 10.1016/j.cld.2017.08.001.
- George Cholankeril; Robert J. Wong; Menghan Hu; Ryan B. Perumpail; Eric R. Yoo; Puneet Puri; Zobair M. Younossi; Stephen A. Harrison; Aijaz Ahmed; Liver Transplantation for Nonalcoholic Steatohepatitis in the US: Temporal Trends and Outcomes. Digestive Diseases and Sciences 2017, 62, 2915-2922, 10.1007/s10620-017-4684-x.
- Day, C.P.; From fat to inflammation.. Gastroenterology 2006, 130, 207-210, https://doi.org/10.1053/j.gastro.2005.11.017.
- Robert J. Wong; Maria Aguilar; Ramsey Cheung; Ryan Perumpail; Stephen A. Harrison; Zobair M. Younossi; Aijaz Ahmed; Nonalcoholic Steatohepatitis Is the Second Leading Etiology of Liver Disease Among Adults Awaiting Liver Transplantation in the United States. Gastroenterology 2015, 148, 547-555, 10.1053/j.gastro.2014.11.039.
- Christopher D Byrne; Giovanni Targher; NAFLD: A multisystem disease. Journal of Hepatology 2015, 62, S47-S64, 10.1016/j.jhep.2014.12.012.
- Marica Meroni; Miriam Longo; Alice Rustichelli; Paola Dongiovanni; Nutrition and Genetics in NAFLD: The Perfect Binomium. International Journal of Molecular Sciences 2020, 21, 2986, 10.3390/ijms21082986.
- Paola Dongiovanni; Erika Paolini; Alberto Corsini; Cesare R. Sirtori; Massimiliano Ruscica; Nonalcoholic fatty liver disease or metabolic dysfunction‐associated fatty liver disease diagnoses and cardiovascular diseases: From epidemiology to drug approaches. European Journal of Clinical Investigation 2021, 51, e13519, 10.1111/eci.13519.
- Paola Dongiovanni; Luca Valenti; Genetics of nonalcoholic fatty liver disease. Metabolism 2015, 65, 1026-1037, 10.1016/j.metabol.2015.08.018.
- Marica Meroni; Miriam Longo; Veronica Erconi; Luca Valenti; Stefano Gatti; Anna Ludovica Fracanzani; Paola Dongiovanni; mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance. Nutrients 2019, 11, 2597, 10.3390/nu11112597.
- Paola Dongiovanni; Marica Meroni; Miriam Longo; Silvia Fargion; Anna Ludovica Fracanzani; miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. International Journal of Molecular Sciences 2018, 19, 3966, 10.3390/ijms19123966.
- Stefan Stender; Julia Kozlitina; Børge G. Nordestgaard; Anne Tybjærg-Hansen; Helen H. Hobbs; Jonathan C. Cohen; Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nature Genetics 2017, 49, 842-847, 10.1038/ng.3855.
- Meroni, M.; Longo, M.; Dongiovanni, P.; Alcohol or Gut Microbiota: Who Is the Guilty?. Int. J. Mol. Sci. 2019, 20, 4568, https://doi.org/10.3390/ijms20184568.
- Marica Meroni; Miriam Longo; Paola Dongiovanni; The Role of Probiotics in Nonalcoholic Fatty Liver Disease: A New Insight into Therapeutic Strategies. Nutrients 2019, 11, 2642, 10.3390/nu11112642.
- Paola Dongiovanni; Luca Valenti; A Nutrigenomic Approach to Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences 2017, 18, 1534, 10.3390/ijms18071534.
- Quentin M. Anstee And Luca Valenti Paola Dongiovanni; Quentin Anstee; Luca Valenti; Genetic Predisposition in NAFLD and NASH: Impact on Severity of Liver Disease and Response to Treatment. Current Pharmaceutical Design 2013, 19, 5219-5238, 10.2174/13816128113199990381.
- Paola Dongiovanni; Stefano Romeo; Luca Valenti; Genetic Factors in the Pathogenesis of Nonalcoholic Fatty Liver and Steatohepatitis. BioMed Research International 2015, 2015, 1-10, 10.1155/2015/460190.
- Veerle Margrethe Diane Struben; Elizabeth Erickson Hespenheide; Stephen H Caldwell; Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. The American Journal of Medicine 2000, 108, 9-13, 10.1016/s0002-9343(99)00315-0.
- Cecilia Lindgren; Laura M. Yerges-Armstrong; Jun Wu; Ruben Hernaez; Lauren J. Kim; Lyle Palmer; Vilmundur Gudnason; Gudny Eiriksdottir; Melissa E. Garcia; Lenore J. Launer; et al.Michael A. NallsJeanne M. ClarkBraxton MitchellAlan ShuldinerJohannah L. ButlerMarta TomasUdo HoffmannShih-Jen HwangJoseph MassaroChristopher J. O'DonnellDushyant V. SahaniVeikko SalomaaEric E. SchadtStephen M. SchwartzDavid S. SiscovickBenjamin F. VoightJohn CarrMary FeitosaTamara B. HarrisCaroline S. FoxAlbert Vernon SmithW. H. Linda KaoJoel N. HirschhornIngrid B. BoreckiNash CrnGIANT ConsortiumMAGIC InvestigatorsGOLD Consortium Genome-Wide Association Analysis Identifies Variants Associated with Nonalcoholic Fatty Liver Disease That Have Distinct Effects on Metabolic Traits. PLoS Genetics 2011, 7, e1001324, 10.1371/journal.pgen.1001324.
- Lynne E. Wagenknecht; Ann L. Scherzinger; Elizabeth R. Stamm; Anthony J. G. Hanley; Jill M. Norris; Yii-Der I. Chen; Michael Bryer-Ash; Steven M. Haffner; Jerome I. Rotter; Correlates and Heritability of Nonalcoholic Fatty Liver Disease in a Minority Cohort. Obesity 2009, 17, 1240-1246, 10.1038/oby.2009.4.
- Jeffrey B. Schwimmer; Manuel A. Celedon; Joel E. Lavine; Rany Salem; Nzali Campbell; Nicholas J. Schork; Masoud Shiehmorteza; Takeshi Yokoo; Alyssa Chavez; Michael S. Middleton; et al.Claude Sirlin Heritability of Nonalcoholic Fatty Liver Disease. Gastroenterology 2009, 136, 1585-1592, 10.1053/j.gastro.2009.01.050.
- Ira R Willner; Bradford Waters; S Raj Patil; Adrian Reuben; Joseph Morelli; Caroline A Riely; Ninety patients with nonalcoholic steatohepatitis: insulin resistance, familial tendency, and severity of disease. American Journal of Gastroenterology 2001, 96, 2957-2961, 10.1016/s0002-9270(01)03229-4.
- Janne Makkonen; Kirsi Pietiläinen; Aila Rissanen; Jaakko Kaprio; Hannele Yki-Järvinen; Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: A study in monozygotic and dizygotic twins. Journal of Hepatology 2009, 50, 1035-1042, 10.1016/j.jhep.2008.12.025.
- Rohit Loomba; Nicholas Schork; Chi-Hua Chen; Ricki Bettencourt; Ana Bhatt; Brandon Ang; Phirum Nguyen; Carolyn Hernandez; Lisa Richards; Joanie Salotti; et al.Steven LinEkihiro SekiKaren E. NelsonClaude B. SirlinDavid Brenner Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015, 149, 1784-1793, 10.1053/j.gastro.2015.08.011.
- Jeffrey Cui; Chi-Hua Chen; Min-Tzu Lo; Nicholas Schork; Ricki Bettencourt; Monica P. Gonzalez; Archana Bhatt; Jonathan Hooker; Katherine Shaffer; Karen E. Nelson; et al.Michelle LongDavid BrennerClaude B. SirlinRohit LoombaFor The Genetics Of Nafld In for the Genetics of NAFLD in Twins Consortium Shared genetic effects between hepatic steatosis and fibrosis: A prospective twin study.. Hepatology 2016, 64, 1547-1558, 10.1002/hep.28674.
- Adam D. Tarnoki; David L. Tarnoki; Pal Bata; Levente Littvay; Janos Osztovits; Gyorgy Jermendy; Kinga Karlinger; Agnes Lannert; Istvan Preda; Robert G. Kiss; et al.Andrea A. MolnarZsolt GaramiGyorgy BaffyViktor Berczi Heritability of non-alcoholic fatty liver disease and association with abnormal vascular parameters: A twin study. Liver International 2012, 32, 1287-1293, 10.1111/j.1478-3231.2012.02823.x.
- Maya Balakrishnan; Fasiha Kanwal; Hashem B. El-Serag; Aaron P. Thrift; Acculturation and Nonalcoholic Fatty Liver Disease Risk Among Hispanics of Mexican Origin: Findings From the National Health and Nutrition Examination Survey. Clinical Gastroenterology and Hepatology 2016, 15, 310-312, 10.1016/j.cgh.2016.09.149.
- Jeffrey D. Browning; Lidia S. Szczepaniak; Robert Dobbins; Pamela Nuremberg; Jay D. Horton; Jonathan C. Cohen; Scott M. Grundy; Helen H. Hobbs; Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387-1395, 10.1002/hep.20466.
- Michael Wayne Fleischman; Matthew Budoff; Ifran Zeb; Ng Li; Temitope Foster; NAFLD prevalence differs among hispanic subgroups: The multi-ethnic study of atherosclerosis. World Journal of Gastroenterology 2014, 20, 4987-93, 10.3748/wjg.v20.i17.4987.
- Guerrero, R.; Vega, G.L.; Grundy, S.M.; Browning, J.D.; Ethnic differences in hepatic steatosis: An insulin resistance paradox?. Hepatology 2009, 49, 791-801, https://doi.org/10.1002/hep.22726.
- P. Dongiovanni; Stefan Stender; A. Pietrelli; Rosellina Margherita Mancina; A. Cespiati; S. Petta; S. Pelusi; P. Pingitore; S. Badiali; M. Maggioni; et al.V. MannistoS. GrimaudoR. M. PipitoneJussi PihlajamäkiA. CraxiM. TaubeL. M.S. CarlssonS. FargionS. RomeoJ. KozlitinaL. Valenti Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. Journal of Internal Medicine 2017, 283, 356-370, 10.1111/joim.12719.
- Di Costanzo, A.; Belardinilli, F.; Bailetti, D.; Sponziello, M.; D’Erasmo, L.; valuation of Polygenic Determinants of Non-Alcoholic Fatty Liver Disease (NAFLD) By a Candidate Genes Resequencing Strategy. Sci. Rep. 2018, 8, 3702, https://doi.org/10.1038/s41598-018-21939-0.
- Krawczyk, M.; Bantel, H.; Rau, M.; Schattenberg, J.M.; Grunhage, F.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Weber, S.N.; et al.et al. Could inherited predisposition drive non-obese fatty liver disease? Results from German tertiary referral centers.. J. Hum. Genet. 2018, 63, 621-626, https://doi.org/10.1038/s10038-018-0420-4.
- Dongiovanni, P.; Rametta, R.; Meroni, M.; Valenti, L.; The role of insulin resistance in nonalcoholic steatohepatitis and liver disease development—A potential therapeutic target?. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 229-242, https://doi.org/10.1586/17474124.2016.1110018.
- Paola Dongiovanni; Marica Meroni; Guido Alessandro Baselli; Giulia Alessandra Bassani; Raffaela Rametta; Alessandro Pietrelli; Marco Maggioni; Federica Facciotti; Valentina Trunzo; Sara Badiali; et al.Silvia FargionStefano GattiLuca Valenti Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clinical Science 2017, 131, 1301-1315, 10.1042/cs20170175.
- Paul Angulo; David E. Kleiner; Sanne Dam-Larsen; Leon A. Adams; Einar S. Bjornsson; Phunchai Charatcharoenwitthaya; Peter R. Mills; Jill C. Keach; Heather D. Lafferty; Alisha Stahler; et al.Svanhildur HaflidadottirFlemming Bendtsen Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389-397.e10, 10.1053/j.gastro.2015.04.043.
- Stuart McPherson; Tim Hardy; Elsbeth Henderson; Alastair Burt; Christopher P. Day; Quentin M. Anstee; Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. Journal of Hepatology 2014, 62, 1148-1155, 10.1016/j.jhep.2014.11.034.
- Serena Pelusi; Salvatore Petta; Chiara Rosso; Vittorio Borroni; Anna Ludovica Fracanzani; Paola Dongiovanni; Antonio Craxi; Elisabetta Bugianesi; Silvia Fargion; Luca Valenti; et al. Renin-Angiotensin System Inhibitors, Type 2 Diabetes and Fibrosis Progression: An Observational Study in Patients with Nonalcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0163069-e0163069, 10.1371/journal.pone.0163069.
- P. Dongiovanni; Luca Valenti; Raffaela Rametta; Ann Daly; Valerio Nobili; Enrico Mozzi; J. B. S. Leathart; Andrea Pietrobattista; Alastair Burt; M. Maggioni; et al.Anna Ludovica FracanzaniE. LattuadaMarco Antonio ZappaG. RoviaroGiulio MarchesiniC. P. DayS. Fargion Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267-273, 10.1136/gut.2009.190801.
- Aya Kitamoto; Takuya Kitamoto; Takahiro Nakamura; Yuji Ogawa; Masato Yoneda; Hideyuki Hyogo; Hidenori Ochi; Seiho Mizusawa; Takato Ueno; Kazuwa Nakao; et al.Akihiro SekineKazuaki ChayamaAtsushi NakajimaKikuko Hotta Association of polymorphisms in GCKR and TRIB1 with nonalcoholic fatty liver disease and metabolic syndrome traits. Endocrine Journal 2014, 61, 683-689, 10.1507/endocrj.ej14-0052.
- Serena Pelusi; Guido Baselli; Alessandro Pietrelli; Paola Dongiovanni; Benedetta Donati; Misti Vanette McCain; Marica Meroni; Anna Ludovica Fracanzani; Renato Romagnoli; Salvatore Petta; et al.Antonio GriecoLuca MieleGiorgio SoardoElisabetta BugianesiSilvia FargionAlessio AghemoRoberta D’AmbrosioChao XingStefano RomeoRaffaele De FrancescoHelen Louise ReevesLuca Vittorio Carlo Valenti Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Scientific Reports 2019, 9, 1-10, 10.1038/s41598-019-39998-2.
- Angelo Baldassare Cefalu'; James Pirruccello; Davide Noto; Stacey Gabriel; Vincenza Valenti; Namrata Gupta; Rossella Spina; Patrizia Tarugi; Sekar Kathiresan; Maurizio R. Averna; et al. A Novel APOB Mutation Identified by Exome Sequencing Cosegregates With Steatosis, Liver Cancer, and Hypocholesterolemia. Arteriosclerosis, Thrombosis, and Vascular Biology 2013, 33, 2021-2025, 10.1161/atvbaha.112.301101.
- Mathilde Di Filippo; Philippe Moulin; Pascal Roy; Marie Elisabeth Samson-Bouma; Sophie Collardeau Frachon; Sabrina Chebel-Dumont; Noël Peretti; Jérôme Dumortier; Fabien Zoulim; Thierry Fontanges; et al.Rossella PariniMiriam RigoldiFrancesca FurlanGrazia ManciniDominique Bonnefont-RousselotEric BruckertJacques SchmitzJean Yves ScoazecSybil CharrièreSylvie Villar-FimbelFrederic GottrandBéatrice DubernDiane DoummarFrancesca JolyMarie Elisabeth Liard-MeillonAlain LachauxAgnès Sassolas Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. Journal of Hepatology 2014, 61, 891-902, 10.1016/j.jhep.2014.05.023.
- Kitt Falk Petersen; Sylvie Dufour; Ali Hariri; Carol Nelson-Williams; Jia Nee Foo; Xian-Man Zhang; James Dziura; Richard P. Lifton; Gerald I. Shulman; Apolipoprotein C3 Gene Variants in Nonalcoholic Fatty Liver Disease. New England Journal of Medicine 2010, 362, 1082-1089, 10.1056/nejmoa0907295.
- Julia Kozlitina; Eric Boerwinkle; Jonathan C. Cohen; Helen H. Hobbs; Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance. Hepatology 2010, 53, 467-474, 10.1002/hep.24072.
- Luca Valenti; Valerio Nobili; Ahmad Al-Serri; Raffaela Rametta; Julian B.S. Leathart; Marco Antonio Zappa; Paola Dongiovanni; Anna Ludovica Fracanzani; Arianna Alterio; Giancarlo Roviaro; et al.Ann DalySilvia FargionChristopher P. Day The APOC3 T-455C and C-482T promoter region polymorphisms are not associated with the severity of liver damage independently of PNPLA3 I148M genotype in patients with nonalcoholic fatty liver. Journal of Hepatology 2011, 55, 1409-1414, 10.1016/j.jhep.2011.03.035.
- Paola Dongiovanni; Marica Meroni; Guido Alessandro Baselli; Rosellina M. Mancina; Massimiliano Ruscica; Miriam Longo; Raffaela Rametta; Annalisa Cespiati; Serena Pelusi; Nicola Ferri; et al.Valeria RanzaniValerio NobiliJussi PihlajamakiAnna Ludovica FracanzaniSara BadialiSalvatore PettaSilvia FargionStefano RomeoJulia KozlitinaLuca Valenti PCSK7 gene variation bridges atherogenic dyslipidemia with hepatic inflammation in NAFLD patients. Journal of Lipid Research 2019, 60, 1144-1153, 10.1194/jlr.p090449.
- Tao Huang; Jinyan Huang; Qibin Qi; Yanping Li; George A. Bray; Jennifer Rood; Frank M. Sacks; Lu Qi; PCSK7Genotype Modifies Effect of a Weight-Loss Diet on 2-Year Changes of Insulin Resistance: The POUNDS LOST Trial. Diabetes Care 2014, 38, 439-444, 10.2337/dc14-0473.
- Yahya Ashraf; Stéphanie Duval; Vatsal Sachan; Rachid Essalmani; Delia Susan‐Resiga; Anna Roubtsova; Josée Hamelin; Stefan Gerhardy; Daniel Kirchhofer; Vincent S. Tagliabracci; et al.Annik PratRobert Scott KissNabil G. Seidah Proprotein convertase 7 (PCSK7) reduces apoA‐V levels. The FEBS Journal 2020, 287, 3565-3578, 10.1111/febs.15212.
- Jonathan Cohen; Alexander Pertsemlidis; Ingrid K Kotowski; Randall Graham; Christine Kim Garcia; Helen H Hobbs; Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nature Genetics 2005, 37, 161-165, 10.1038/ng1509.
- Massimiliano Ruscica; Nicola Ferri; Chiara Macchi; Marica Meroni; Claudia Lanti; Chiara Ricci; Marco Maggioni; Anna Ludovica Fracanzani; Sara Badiali; Silvia Fargion; et al.Paolo MagniLuca ValentiPaola Dongiovanni Liver fat accumulation is associated with circulating PCSK9. Annals of Medicine 2016, 48, 384-391, 10.1080/07853890.2016.1188328.
- Mark Trinder; Gordon A. Francis; Liam R. Brunham; Association of Monogenic vs Polygenic Hypercholesterolemia With Risk of Atherosclerotic Cardiovascular Disease. JAMA Cardiology 2020, 5, 390-399, 10.1001/jamacardio.2019.5954.
- Philippe Costet; Bertrand Cariou; Gilles Lambert; Florent Lalanne; Bernard Lardeux; Anne-Laure Jarnoux; Aldo Grefhorst; Bart Staels; Michel Krempf; Hepatic PCSK9 Expression Is Regulated by Nutritional Status via Insulin and Sterol Regulatory Element-binding Protein 1c. Journal of Biological Chemistry 2006, 281, 6211-6218, 10.1074/jbc.m508582200.
- Ingrid K. Kotowski; Alexander Pertsemlidis; Amy Luke; Richard S. Cooper; Gloria L. Vega; Jonathan C. Cohen; Helen H. Hobbs; A Spectrum of PCSK9 Alleles Contributes to Plasma Levels of Low-Density Lipoprotein Cholesterol. The American Journal of Human Genetics 2006, 78, 410-422, 10.1086/500615.
- Stefania Grimaudo; Stefano Bartesaghi; Raffaela Rametta; Fabio Marra; Rosellina Margherita Mancina; Jussi Pihlajamäki; Dorota Kakol‐Palm; Anne‐Christine Andréasson; Paola Dongiovanni; Anna Ludovica Fracanzani; et al.Giulia LoriVille MännistöGiovanni PellegriniMohammad Bohlooly‐YGrazia PennisiRosaria Maria PipitoneRocco SpagnuoloAntonio CraxìDaniel LindénLuca ValentiStefano RomeoSalvatore Petta PCSK9 rs11591147 R46L loss‐of‐function variant protects against liver damage in individuals with NAFLD. Liver International 2020, 41, 321-332, 10.1111/liv.14711.
- Monica Gomaraschi; Anna Ludovica Fracanzani; Paola Dongiovanni; Chiara Pavanello; Eleonora Giorgio; Lorenzo Da Dalt; Giuseppe Danilo Norata; Laura Calabresi; Dario Consonni; Rosa Lombardi; et al.Adriana BranchiSilvia Fargion Lipid accumulation impairs lysosomal acid lipase activity in hepatocytes: Evidence in NAFLD patients and cell cultures. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2019, 1864, 158523, 10.1016/j.bbalip.2019.158523.
- Željko Reiner; Ornella Guardamagna; Devaki Nair; Handrean Soran; Kees Hovingh; Stefano Bertolini; Simon Jones; Marijana Ćorić; Sebastiano Calandra; John Hamilton; et al.Terence EagletonEmilio Ros Lysosomal acid lipase deficiency – An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21-30, 10.1016/j.atherosclerosis.2014.04.003.
- Hernando Lyons; Eleftherios Vouyoukas; Martha Higgins; James J. Maciejko; Clinical and Histologic Liver Improvement in Siblings With Lysosomal Acid Lipase Deficiency After Enzyme Replacement. Journal of Pediatric Gastroenterology & Nutrition 2020, 70, 635-639, 10.1097/mpg.0000000000002671.
- Vĕra Malinová; Manisha Balwani; Reena Sharma; Jean‐Baptiste Arnoux; John Kane; Chester B. Whitley; Sachin Marulkar; Florian Abel; Sebelipase alfa for lysosomal acid lipase deficiency: 5‐year treatment experience from a phase 2 open‐label extension study. Liver International 2020, 40, 2203-2214, 10.1111/liv.14603.
- A. Auinger; Luca Valenti; M. Pfeuffer; U. Helwig; J. Herrmann; Anna Ludovica Fracanzani; P. Dongiovanni; S. Fargion; J. Schrezenmeir; D. Rubin; et al. A Promoter Polymorphism in the Liver-specific Fatty Acid Transport Protein 5 is Associated with Features of the Metabolic Syndrome and Steatosis. Hormone and Metabolic Research 2010, 42, 854-859, 10.1055/s-0030-1267186.
- Luca Valenti; Benedetta Maria Motta; Anna Alisi; Rita Sartorelli; Giulia Buonaiuto; Paola Dongiovanni; Raffaela Rametta; Serena Pelusi; Silvia Fargion; Valerio Nobili; et al. LPIN1 rs13412852 Polymorphism in Pediatric Nonalcoholic Fatty Liver Disease. Journal of Pediatric Gastroenterology & Nutrition 2012, 54, 588-593, 10.1097/mpg.0b013e3182442a55.
- Claire S. Faulkner; Collin M. White; Vijay H. Shah; Loretta L. Jophlin; A single nucleotide polymorphism of PLIN2 is associated with nonalcoholic steatohepatitis and causes phenotypic changes in hepatocyte lipid droplets: A pilot study. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2020, 1865, 158637-158637, 10.1016/j.bbalip.2020.158637.
- Joëlle Magné; Anna Aminoff; Jeanna Perman Sundelin; Maria Nastase Mannila; Peter Gustafsson; Kjell Hultenby; Annika Wernerson; Greta Bauer; Laura Listenberger; Matt J. Neville; et al.Fredrik KarpeJan BorénEwa Ehrenborg The minor allele of the missense polymorphism Ser251Pro in perilipin 2 (PLIN2) disrupts an α‐helix, affects lipolysis, and is associated with reduced plasma triglyceride concentration in humans. The FASEB Journal 2013, 27, 3090-3099, 10.1096/fj.13-228759.
- Christina Drevinge; Knut T. Dalen; Maria Nastase Mannila; Margareta Scharin Täng; Marcus Ståhlman; Martina Klevstig; Annika Lundqvist; Ismena Mardani; Fred Haugen; Per Fogelstrand; et al.Martin AdielsJorge Asin-CayuelaCharlotte EkestamJesper R. GådinYun K. LeeHilde NebbSara SvedlundBengt R. JohanssonLillemor Mattsson HulténStefano RomeoBjörn RedforsElmir OmerovicMax LevinLi-Ming GanPer ErikssonLinda AnderssonEwa EhrenborgAlan R. KimmelJan BorénMalin C. Levin Perilipin 5 is protective in the ischemic heart. International Journal of Cardiology 2016, 219, 446-454, 10.1016/j.ijcard.2016.06.037.
- Kenta Kuramoto; Tomoo Okamura; Tomohiro Yamaguchi; Tomoe Y. Nakamura; Shigeo Wakabayashi; Hidetaka Morinaga; Masatoshi Nomura; Toshihiko Yanase; Kinya Otsu; Nobuteru Usuda; et al.Shigenobu MatsumuraKazuo InoueTohru FushikiYumiko KojimaTakeshi HashimotoFumie SakaiFumiko HiroseTakashi Osumi Perilipin 5, a Lipid Droplet-binding Protein, Protects Heart from Oxidative Burden by Sequestering Fatty Acid from Excessive Oxidation. Journal of Biological Chemistry 2012, 287, 23852-23863, 10.1074/jbc.m111.328708.
- Yu-Cheng Lin; Pi-Feng Chang; Hsueh-Fang Lin; Kevin Liu; Mei Hwei Chang; Yen-Hsuan Ni; Variants in the autophagy-related gene IRGM confer susceptibility to non-alcoholic fatty liver disease by modulating lipophagy. Journal of Hepatology 2016, 65, 1209-1216, 10.1016/j.jhep.2016.06.029.
- Kristin Schwerbel; Anne Kamitz; Natalie Krahmer; Nicole Hallahan; Markus Jähnert; Pascal Gottmann; Sandra Lebek; Tanja Schallschmidt; Danny Arends; Fabian Schumacher; et al.Burkhard KleuserTom HaltenhofFlorian HeydSofiya GanchevaKarl W. BromanMichael RodenHans-Georg JoostAlexandra ChadtHadi Al-HasaniHeike VogelWenke JonasAnnette Schürmann Immunity-related GTPase induces lipophagy to prevent excess hepatic lipid accumulation. Journal of Hepatology 2020, 73, 771-782, 10.1016/j.jhep.2020.04.031.
- Marra, F.; Bertolani, C.; Adipokines in liver diseases.. Hepatology 2009, 50, 957-969, https://doi.org/10.1002/hep.23046.
- Luca Miele; Venanzio Valenza; Giuseppe La Torre; Massimo Montalto; Giovanni Cammarota; Riccardo Ricci; Roberta Mascianà; Alessandra Forgione; Maria L. Gabrieli; Germano Perotti; et al.Fabio M. VecchioGianlodovico RapacciniGiovanni GasbarriniChris P. DayAntonio Grieco Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877-1887, 10.1002/hep.22848.
- Manfred Bilzer; Frigga Roggel; Alexander L. Gerbes; Role of Kupffer cells in host defense and liver disease. Liver International 2006, 26, 1175-1186, 10.1111/j.1478-3231.2006.01342.x.
- Mohammed Eslam; the International Liver Disease Genetics Consortium (ILDGC); Duncan McLeod; Kebitsaone Simon Kelaeng; Alessandra Mangia; Thomas Berg; Khaled Thabet; William Irving; Gregory J Dore; David Sheridan; et al.Henning GrønbækMaria Lorena AbateRune HartmannElisabetta BugianesiUlrich SpenglerAngela RojasDavid R BoothMartin WeltmanLindsay MollisonWendy ChengStephen RiordanHema MahajanJanett FischerJacob NattermannMark W DouglasChristopher LiddleElizabeth PowellManuel Romero-GomezJacob George IFN-λ3, not IFN-λ4, likely mediates IFNL3–IFNL4 haplotype–dependent hepatic inflammation and fibrosis. Nature Genetics 2017, 49, 795-800, 10.1038/ng.3836.
- Salvatore Petta; Stefania Grimaudo; Calogero Cammà; Daniela Cabibi; Vito Di Marco; Giusalba Licata; Rosaria Maria Pipitone; Antonio Craxì; IL28B and PNPLA3 polymorphisms affect histological liver damage in patients with non-alcoholic fatty liver disease. Journal of Hepatology 2012, 56, 1356-1362, 10.1016/j.jhep.2012.01.007.
- Mohammed Eslam; the International Hepatitis C Genetics Consortium (IHCGC); Ahmed M. Hashem; Reynold Leung; Manuel Romero-Gómez; Thomas Berg; Gregory J. Dore; Henry L.K. Chan; William Irving; David Sheridan; et al.Maria L. AbateLeon A. AdamsAlessandra MangiaMartin WeltmanElisabetta BugianesiUlrich SpenglerOlfat ShakerJanett FischerLindsay MollisonWendy ChengElizabeth PowellJacob NattermannStephen RiordanDuncan McLeodNicola J. ArmstrongMark W. DouglasChristopher LiddleDavid R. BoothJacob GeorgeGolo Ahlenstiel Interferon-λ rs12979860 genotype and liver fibrosis in viral and non-viral chronic liver disease. Nature Communications 2015, 6, 6422, 10.1038/ncomms7422.
- Mohammed Eslam; Ahmed M. Hashem; Manuel Romero-Gómez; Thomas Berg; Gregory J. Dore; Alessandra Mangia; Henry Lik Yuen Chan; William Irving; David Sheridan; Maria Lorena Abate; et al.Leon A. AdamsMartin WeltmanElisabetta BugianesiUlrich SpenglerOlfat ShakerJanett FischerLindsay MollisonWendy ChengJacob NattermannStephen RiordanLuca MieleKebitsaone Simon KelaengJavier AmpueroGolo AhlenstielDuncan McLeodElizabeth PowellChristopher LiddleMark W. DouglasDavid R. BoothJacob George FibroGENE: A gene-based model for staging liver fibrosis. Journal of Hepatology 2016, 64, 390-398, 10.1016/j.jhep.2015.11.008.
- Petta, S.; Valenti, L.; Interferon lambda 4 rs368234815 TT > δG variant is associated with liver damage in patients with nonalcoholic fatty liver disease.. J. Hepatol. 2017, 66, 1885-1893, https://doi.org/10.1002/hep.29395.
- Petta, S.; Valenti, L.; Svegliati-Baroni, G.; Ruscica, M.; Pipitone, R.M.; Dongiovanni, P.; Rychlicki, C.; Ferri, N.; Cammà, C.; Fracanzani, A.L.; et al.et al. Fibronectin Type III Domain-Containing Protein 5 rs3480 A > G Polymorphism, Irisin, and Liver Fibrosis in Patients With Nonalcoholic Fatty Liver Disease.. J. Clin. Endocrinol. Metab. 2017, 102, 2660-2669, https://doi.org/10.1210/jc.2017-00056.
- Mayada Metwally; Ali Bayoumi; Manuel Romero-Gomez; Khaled Thabet; Miya John; Leon A. Adams; Xiaoqi Huo; Rocio Aller; Carmelo García-Monzón; María Teresa Arias-Loste; et al.Elisabetta BugianesiLuca MieleRocío Gallego-DuránJanett FischerThomas BergChristopher LiddleLiang QiaoJacob GeorgeMohammed Eslam A polymorphism in the Irisin-encoding gene (FNDC5) associates with hepatic steatosis by differential miRNA binding to the 3′UTR. Journal of Hepatology 2018, 70, 494-500, 10.1016/j.jhep.2018.10.021.
- Alexis Gorden; Rongze Yang; Laura M. Yerges-Armstrong; Kathleen A. Ryan; Elizabeth Speliotes; Ingrid B. Borecki; Tamara B. Harris; Xin Chu; G. Craig Wood; Christopher D. Still; et al.Alan R. ShuldinerGlenn S. GerhardGOLD Consortium Genetic Variation at NCAN Locus Is Associated with Inflammation and Fibrosis in Non-Alcoholic Fatty Liver Disease in Morbid Obesity. Human Heredity 2013, 75, 34-43, 10.1159/000346195.
- Cai, W.; Weng, D.H.; Yan, P.; Lin, Y.T.; Dong, Z.H.; Mailamuguli; Yao, H.; Genetic polymorphisms associated with nonalcoholic fatty liver disease in Uyghur population: A case-control study and meta-analysis.. Lipids Health Dis. 2019, 18, 14, https://doi.org/10.1186/s12944-018-0877-3.
- Claudia Cicione; Chiara Degirolamo; Antonio Moschetta; Emerging role of fibroblast growth factors 15/19 and 21 as metabolic integrators in the liver. Hepatology 2012, 56, 2404-2411, 10.1002/hep.25929.
- Dongiovanni, P.; Crudele, A.; Panera, N.; Romito, I.; Meroni, M.; De Stefanis, C.; Palma, A.; Comparcola, D.; Fracanzani, A.L.; Miele, L.; et al.et al. beta-Klotho gene variation is associated with liver damage in children with NAFLD.. J. Hepatol. 2020, 72, 411-419, https://doi.org/10.1016/j.jhep.2019.10.011.
- Kathleen Markan; Meghan C. Naber; Sarah M. Small; Lila Peltekian; Rachel L. Kessler; Matthew J. Potthoff; FGF21 resistance is not mediated by downregulation of beta-klotho expression in white adipose tissue. Molecular Metabolism 2017, 6, 602-610, 10.1016/j.molmet.2017.03.009.
- Dushay, J.; Chui, P.C.; Gopalakrishnan, G.S.; Varela-Rey, M.; Crawley, M.; Fisher, F.M.; Badman, M.K.; Martinez-Chantar, M.L.; Maratos-Flier, E.; Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease.. Gastroenterology 2010, 139, 456-463, https://doi.org/10.1053/j.gastro.2010.04.054.
- Javier Gómez-Ambrosi; Jose Miguel Gallego-Escuredo; Victoria Catalan; Amaia Rodríguez; Pere Domingo; Rafael Moncada; Víctor Valentí; Javier Salvador; Marta Giralt; Francesc Villarroya; et al.Gema Frühbeck FGF19 and FGF21 serum concentrations in human obesity and type 2 diabetes behave differently after diet- or surgically-induced weight loss. Clinical Nutrition 2016, 36, 861-868, 10.1016/j.clnu.2016.04.027.
- Luca Miele; Gary Beale; Gillian Patman; Valerio Nobili; Julian Leathart; Antonio Grieco; Marilena Abate; Scott L. Friedman; Goutham Narla; Elisabetta Bugianesi; et al.Christopher P. DayHelen L. Reeves The Kruppel-Like Factor 6 Genotype Is Associated With Fibrosis in Nonalcoholic Fatty Liver Disease. Gastroenterology 2008, 135, 282-291.e1, 10.1053/j.gastro.2008.04.004.
- Raffaela Rametta; Marica Meroni; Paola Dongiovanni; From Environment to Genome and Back: A Lesson from HFE Mutations. International Journal of Molecular Sciences 2020, 21, 3505, 10.3390/ijms21103505.
- Salvatore Petta; Luca Valenti; Fabio Marra; Stefania Grimaudo; Claudio Tripodo; Elisabetta Bugianesi; Calogero Cammà; Andrea Cappon; Vito Di Marco; Giovanni Di Maira; et al.Paola DongiovanniRaffaela RamettaAlessandro GulinoEnrico MozziEmanuele OrlandoMarco MaggioniRosaria Maria PipitoneSilvia FargionAntonio Craxì MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. Journal of Hepatology 2016, 64, 682-690, 10.1016/j.jhep.2015.10.016.
- S Rüeger; P-Y Bochud; J-F Dufour; B Müllhaupt; D Semela; M H Heim; D Moradpour; A Cerny; R Malinverni; D R Booth; et al.V SuppiahJ GeorgeL ArgiroP HalfonM BourlièreA H TalalI M JacobsonE PatinB NalpasT PoynardS PolL AbelZ KutalikF Negro Impact of common risk factors of fibrosis progression in chronic hepatitis C. Gut 2014, 64, 1605-1615, 10.1136/gutjnl-2014-306997.
- Bishuang Cai; Paola Dongiovanni; Kathleen E. Corey; Xiaobo Wang; Igor O. Shmarakov; Ze Zheng; Canan Kasikara; Viralkumar Davra; Marica Meroni; Raymond T. Chung; et al.Carla V. RothlinRobert F. SchwabeWilliam S. BlanerRaymond B. BirgeLuca ValentiIra Tabas Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metabolism 2020, 31, 406-421.e407, 10.1016/j.cmet.2019.11.013.
- José A. Nicolás-Ávila; Ana V. Lechuga-Vieco; Lorena Esteban-Martínez; María Sánchez-Díaz; Elena Díaz García; Demetrio J. Santiago; Andrea Rubio-Ponce; Jackson LiangYao Li; Akhila Balachander; Juan A. Quintana; et al.Raquel Martínez-De-MenaBeatriz Castejón-VegaAndrés Pun-GarcíaPaqui G. TravésElena Bonzón-KulichenkoFernando García-MarquésLorena CussóNoelia A-GonzálezAndrés González-GuerraMarta Roche-MolinaSandra Martin-SalamancaGeorgiana CrainiciucGabriela GuzmánJagoba LarrazabalElías Herrero-GalánJorge Alegre-CebolladaGreg LemkeCarla V. RothlinLuis Jesús Jimenez-BorregueroGuillermo ReyesAntonio CastrilloManuel DescoPura Muñoz-CánovesBorja IbáñezMiguel TorresLai Guan NgSilvia G. PrioriHéctor BuenoJesús VázquezMario D. CorderoJuan A. BernalJosé A. EnríquezAndrés Hidalgo A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94-109.e23, 10.1016/j.cell.2020.08.031.
- Aloysious Aravinthan; George Mells; Michael Allison; Julian Leathart; Anna Kotronen; Hannele Yki-Järvinen; Ann K Daly; Christopher P Day; Quentin M. Anstee; Graeme Alexander; et al. Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease. Function of a membrane-embedded domain evolutionarily multiplied in the GPI lipid anchor pathway proteins PIG-B, PIG-M, PIG-U, PIG-W, PIG-V, and PIG-Z 2014, 13, 1489-1494, 10.4161/cc.28471.
- Rodrigo T. Calado; Joshua A. Regal; David E. Kleiner; David S. Schrump; Nathan R. Peterson; Verònica Pons; Stephen J. Chanock; Peter M. Lansdorp; Neal S. Young; A Spectrum of Severe Familial Liver Disorders Associate with Telomerase Mutations. PLoS ONE 2009, 4, e7926, 10.1371/journal.pone.0007926.
- Meroni, M.; Longo, M.; Paolini, E.; Alisi, A.; Miele, L.; De Caro, E.R.; Pisano, G.; Maggioni, M.; Soardo, G.; Valenti, L.V.; et al.et al. The rs599839 A > G Variant Disentangles Cardiovascular Risk and Hepatocellular Carcinoma in NAFLD Patients.. Cancers (Basel) 2021, 13, 1783, https://doi.org/10.3390/cancers13081783.
- Paola Dongiovanni; Marica Meroni; Salvatore Petta; Miriam Longo; Anna Alisi; Giorgio Soardo; Luca Valenti; Luca Miele; Stefania Grimaudo; Grazia Pennisi; et al.Grieco AntonioDario ConsonniSilvia FargionAnna Ludovica Fracanzani Neurotensin up-regulation is associated with advanced fibrosis and hepatocellular carcinoma in patients with MAFLD. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2020, 1865, 158765, 10.1016/j.bbalip.2020.158765.
- Pessayre, D.; Fromenty, B.; NASH: A mitochondrial disease.. J. Hepatol. 2005, 42, 928-940, https://doi.org/10.1016/j.jhep.2005.03.004.
- Abdellah Mansouri; Charles-Henry Gattolliat; Tarik Asselah; Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629-647, 10.1053/j.gastro.2018.06.083.
- Chikako Namikawa; Zhang Shu-Ping; John Raynor Vyselaar; Yasuko Nozaki; Yoshihisa Nemoto; Masafumi Ono; Naoaki Akisawa; Toshiji Saibara; Makoto Hiroi; Hideaki Enzan; et al.Saburo Onishi Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. Journal of Hepatology 2004, 40, 781-786, 10.1016/j.jhep.2004.01.028.
- Ahmad Al-Serri; Quentin M. Anstee; Luca Valenti; Valerio Nobili; Julian B.S. Leathart; Paola Dongiovanni; Julia Patch; Anna Ludovica Fracanzani; Silvia Fargion; Christopher P. Day; et al.Ann Daly The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. Journal of Hepatology 2011, 56, 448-454, 10.1016/j.jhep.2011.05.029.
- Fares, R.; Petta, S.; Lombardi, R.; Grimaudo, S.; Dongiovanni, P.; Pipitone, R.; Rametta, R.; Fracanzani, A.L.; Mozzi, E.; Craxì, A.; et al.et al. The UCP2 -866 G > A promoter region polymorphism is associated with nonalcoholic steatohepatitis. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 1574-1580, https://doi.org/10.1111/liv.12707.
- G Serviddio; F Bellanti; R Tamborra; T Rollo; N Capitanio; A D Romano; J Sastre; G Vendemiale; E Altomare; Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008, 57, 957-965, 10.1136/gut.2007.147496.
- D. A. de Luis; Role of -55CT polymorphism of UCP3 gene on non alcoholic fatty liver disease and insulin resistance in patients with obesity. Nutr. Hosp. 2010, 25, 572-5576, 10.3305/NH.2010.25.4.4484.
- Lei Zhong; Raul Mostoslavsky; Fine Tuning Our Cellular Factories: Sirtuins in Mitochondrial Biology. Cell Metabolism 2011, 13, 621-626, 10.1016/j.cmet.2011.05.004.
- Chuanhui Dong; David Della-Morte; Liyong Wang; Digna Cabral; Ashley Beecham; Mark S. McClendon; Corneliu Luca; Susan H. Blanton; Ralph L. Sacco; Tatjana Rundek; et al. Association of the Sirtuin and Mitochondrial Uncoupling Protein Genes with Carotid Plaque. PLoS ONE 2011, 6, e27157, 10.1371/journal.pone.0027157.
- Connor A. Emdin; Mary E. Haas; Amit V. Khera; Krishna Aragam; Mark Chaffin; Derek Klarin; George Hindy; Lan Jiang; Wei-Qi Wei; Qiping Feng; et al.Juha KarjalainenAki HavulinnaTuomo KiiskinenAlexander BickDiego ArdissinoJames G. WilsonHeribert SchunkertRuth McPhersonHugh WatkinsRoberto ElosuaMatthew J. BownNilesh J. SamaniUsman BaberJeanette ErdmannNamrata GuptaJohn DaneshDanish SaleheenKyong-Mi ChangMarijana VujkovicBen VoightScott DamrauerJulie LynchDavid KaplanMarina SerperPhilip TsaoJosep MercaderCraig HanisMark DalyJoshua DennyStacey GabrielSekar KathiresanMillion Veteran Program A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genetics 2020, 16, e1008629, 10.1371/journal.pgen.1008629.
- Hamish Innes; Stephan Buch; Sharon Hutchinson; Indra Neil Guha; Joanne R. Morling; Elleanor Barnes; Will Irving; Ewan Forrest; Vincent Pedergnana; David Goldberg; et al.Esther AspinallStephan BarclayPeter C. HayesJohn DillonHans Dieter NischalkePhilipp LutzUlrich SpenglerJanett FischerThomas BergMario BroschFlorian EyerChristian DatzSebastian MuellerTeresa PeccerellaPierre DeltenreAstrid MarotMichael SoykaAndrew McQuillinMarsha Y. MorganJochen HampeFelix Stickel Genome-Wide Association Study for Alcohol-Related Cirrhosis Identifies Risk Loci in MARC1 and HNRNPUL1. Gastroenterology 2020, 159, 1276-1289.e7, 10.1053/j.gastro.2020.06.014.
- Panu K. Luukkonen; Anne Juuti; Henna Sammalkorpi; Anne K. Penttilä; Matej Orešič; Tuulia Hyötyläinen; Johanna Arola; Marju Orho-Melander; Hannele Yki-Järvinen; MARC1 variant rs2642438 increases hepatic phosphatidylcholines and decreases severity of non-alcoholic fatty liver disease in humans. Journal of Hepatology 2020, 73, 725-726, 10.1016/j.jhep.2020.04.021.
- Jake Mann; Maik Pietzner; Laura B Wittemans; Emmanuela De Lucia Rolfe; Nicola D Kerrison; Fumiaki Imamura; Nita Forouhi; Eric Fauman; Michael E Allison; Jules L Griffin; et al.Albert KoulmanNicholas WarehamClaudia Langenberg Insights into genetic variants associated with NASH-fibrosis from metabolite profiling.. Hum. Mol. Genet. 2020, 29, 3451–3463, 10.17863/cam.55211.
- Carolin V. Schneider; Kai Markus Schneider; Donna M. Conlon; Joseph Park; Marijana Vujkovic; Inuk Zandvakili; Yi-An Ko; Christian Trautwein; Rotonya M. Carr; Pavel Strnad; et al.Christoph A. ThaissDaniel J. Rader A genome-first approach to mortality and metabolic phenotypes in MTARC1 p.Ala165Thr (rs2642438) heterozygotes and homozygotes. Med 2021, 2, 851-863.e853, 10.1016/j.medj.2021.04.011.

