Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Soraya Taleb | + 2485 word(s) | 2485 | 2021-09-24 04:50:06 | | | |
| 2 | Rita Xu | Meta information modification | 2485 | 2021-10-13 06:00:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Taleb, S. Tryptophan Metabolism in Cardiovascular Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/14983 (accessed on 23 July 2026).
Taleb S. Tryptophan Metabolism in Cardiovascular Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/14983. Accessed July 23, 2026.
Taleb, Soraya. "Tryptophan Metabolism in Cardiovascular Diseases" Encyclopedia, https://encyclopedia.pub/entry/14983 (accessed July 23, 2026).
Taleb, S. (2021, October 12). Tryptophan Metabolism in Cardiovascular Diseases. In Encyclopedia. https://encyclopedia.pub/entry/14983
Taleb, Soraya. "Tryptophan Metabolism in Cardiovascular Diseases." Encyclopedia. Web. 12 October, 2021.
Copy Citation
Cardiovascular disease (CVD) is one of the major causes of mortality worldwide. Inflammation is the underlying common mechanism involved in CVD. It has been recently related to amino acid metabolism, which acts as a critical regulator of innate and adaptive immune responses. Among different metabolites that have emerged as important regulators of immune and inflammatory responses, tryptophan (Trp) metabolites have been shown to play a pivotal role in CVD.
tryptophan
tryptophan catabolism
kynurenine
IDO
cardiovascular disease
1. Introduction
Cardiovascular disease (CVD), including myocardial infarction (MI) and stroke, constitutes the leading cause of death worldwide. Atherosclerosis is the underlying cause of the majority of CVD. Despite the use of cholesterol-lowering therapies to reduce atherosclerosis, nearly 18 million people die each year due to CVD [1]. Given the high prevalence of obesity in developing countries, the global incidence of CVD is predicted to increase and constitute the greatest public health problem in the world.
Atherosclerosis is a chronic inflammatory disease affecting both large and medium arteries initiated by increased endothelial cell (EC) permeability and intimal low-density lipoprotein (LDL) cholesterol accumulation [2]. Over years, atherosclerotic plaque overgrowth can lead to plaque instability and a high probability of rupture. The disruption of the plaque surface and exposure of its thrombogenic content to the luminal blood flow result in thromboembolism, which may occur in one or more coronary arteries, contributing to their occlusion [2]. Therefore, blood oxygen and nutrient deprivation result in ischemia, and consequently myocardial cell death, contributing to acute MI most commonly occurring in the left ventricle (LV) [3][4]. MI is therefore considered as the first and most severe clinical manifestation of atherosclerosis.
Inflammation was identified as a key driver of atherothrombotic events [5][6][7]. On this subject, the authors of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) trial showed that targeting interleukin-1β (IL-1β) improved CV outcome in patients with MI history, which would confirm the proof of concept of the role of inflammation in the causal pathway of atherothrombosis disease [8]. This was followed by the Low-Dose Colchicine after Myocardial Infarction (COLCOT) trial which revealed that non-selective inhibition of inflammation using colchicine significantly reduced atherosclerosis-related events [9].
Several actors can contribute to inflammatory-associated response which include modifiable and non-modifiable risk factors. Among modifiable risk factors, diet and particularly its composition has been suggested to significantly impact inflammation. In this regard, some amino acids (AAs) metabolic pathways have become critical checkpoints for controlling inflammatory-related mechanisms. For instance, branched-chain AAs (leucine, isoleucine, and valine) have been shown to promote EC dysfunction through increased reactive oxygen species (ROS) generation and inflammation [10]. Likewise, tryptophan (Trp) metabolites have been shown to be closely related to inflammation and thereby suggested to be involved in CVD [11]. In that respect, some systemic Trp catabolites were associated with worse CVD outcome and have been considered as important predictors of major acute coronary events [12][13][14][15], as well as biomarkers of adverse LV remodeling following MI [16].
2. Trp Absorption and Catabolism
Trp was discovered by the English chemist Sir Frederick Gowland Hopkins in 1901 [17]. It is one of the nine essential amino acids that cannot be synthesized endogenously and is therefore supplied by the diet. Daily consumption of Trp is about 900–1000 mg, a level which is high considering the WHO recommendation of about 4 mg/kg (~300 mg for an adult). Trp is found in protein-rich foods, such as meat, fish, eggs, milk, cheese, dairy foods, beans, nuts, pumpkin, sesame seeds, turkey, tofu, soy, and chocolate [18].
Following food digestion, Trp absorption occurs through B0AT1 and TAT1, located respectively in the apical and basolateral membranes [19][20]. Some intestinal Trp enters portal and then systemic circulation after absorption and the remaining Trp is locally transformed, notably by gut microbiota into various catabolites.
Trp passes into the hepatic portal system before the unused fraction is released into the bloodstream and delivered to peripheral tissues. In addition to protein synthesis, which represents a small portion (1%), Trp mostly undergoes degradation, a process known as Trp catabolism (Figure 1). Trp-derived metabolites are generated through three known pathways orchestrated by tissue-specific enzymes. The Kynurenine pathway (KP) accounts for ~95% of ingested Trp [21]. Under normal physiological conditions, the first and rate limiting step of Trp catabolism is mostly catalyzed by tryptophan 2, 3-dioxygenase (TDO), mainly expressed in the liver. Its activity is constitutive but could be increased by glucocorticoids and circulating levels of Trp [22]. A minimal contribution is also possible due to the extrahepatic cytosolic enzyme, IDO, and to a lesser extent by its isoform, the indoleamine 2, 3-dioxygenase 2 (IDO2). As a result of TDO, IDO, or IDO2 activity, N-formylKynurenine (NFK) is generated and hydrolyzed to Kyn by NFK formidase. Kyn is then catabolized to several metabolites via the action of multiple enzymes. Kyn is converted to anthranilic acid (AA) by kynureninase, or kynurenic acid (Kna) by kynurenine aminotransferase (KAT), and to 3-hydroxykynurenine (3-OHKyn) by kynurenine monooxygenase (KMO). Then, 3-OHKyn is metabolized into 3-hydroxyanthranilic acid (3-HAA) by kynureninase. As final steps, quinolinic acid (Quina) is generated, and thereafter nicotinamide adenine dinucleotide (NAD), which plays an essential role in energy metabolism. The extrahepatic KP is minimal (<2%) under homeostasis, but becomes quantitatively more significant under inflammatory conditions, when the activity of IDO exceeds that of hepatic TDO [22].
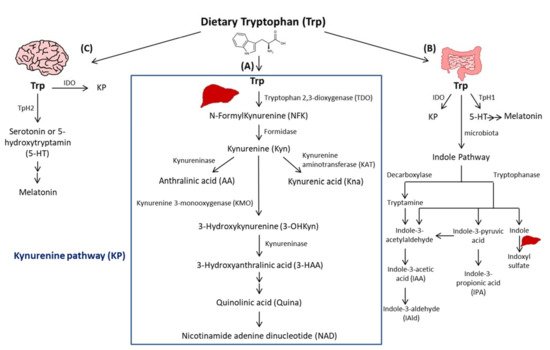
Figure 1. Simplified illustration of the tryptophan degradation in different peripheral organs. Tryptophan (Trp) is predominantly converted into kynurenine (Kyn) pathway (KP) by Tryptophan 2, 3-dioxygenase (TDO) in the liver (A) and by indoleamine 2, 3-dioxygenase 1 (IDO) in extrahepatic organs. In the gastrointestinal tract, a small amount of Trp is converted by gut microbiota through the action of the enzymes tryptophanase and decaboxylase, into indole and its derivatives and into tryptamine (B). Indole metabolites could be converted in the liver into indoxyl sulfate. Another fraction of Trp is converted through Trp hydroxylase 1 (TpH 1) into serotonin in the gut (B) and through Tph2 in the brain (C) then to melatonin.
The second pathway of Trp degradation is the serotonin pathway (~1–2%) mediated by Trp hydroxylase (TPH) that results in the generation of serotonin or 5-hydroxytryptamin (5-HT), the precursor of melatonin [21][23]. The two enzymes involved in Trp hydroxylation are TPH1, mainly expressed in the enterochromaffin cells in the gut [24], and the TPH2 preferentially expressed in the brain [25]. TPH1, whose activity is in part modulated by gut microbiota [26], is responsible for the generation of more than 90% of 5-HT of the body [26]. The serotonin from the gut can enter the circulation or the intestinal lumen to exert its action on almost all the organs. It regulates gut motility, vascular tone, primary hemostasis, and cell-mediated immune response [27]. In the central nervous system, 5-HT generated due to TPH2, controls mood, sleep, anxiety, and food intake. Interestingly, since deregulated TPH2 activity has been implicated in psychiatric disorders, it has been considered as a therapeutic target for these diseases [28]. Of note, serotonin reuptake inhibitors (SSRIs) are considered the most used anti-depressant drugs. Besides their role in blocking 5-HT reuptake transporters in the brain, SSRIs are responsible for 5-HT storage depletion in the platelets, which are the major storage cells for peripheral 5-HT. Melatonin results from 5-HT catabolism in mainly pineal gland in which the peak of its production and release happens during night. It is known to regulate sleep and circadian rhythms, and to act as a free radical scavenger as well as a suppressor of inflammation [23].
In the gastrointestinal tract, Trp is mostly converted in the host to Kyn by IDO, and to a lesser extent to 5-HT, and to indole metabolites by the microbiota [21]. Trp catabolism has a major role in fine-tuning intestinal physiology that may affect peripheral health [29]. In this context, it has been shown that aberrant Trp metabolism and gut microbiota interplay contributes to immune response activation in experimental systemic lupus erythematosus [30]. Consistently, mice lacking dietary Trp displayed an impaired intestinal immunity and microbial dysbiosis [31]. On the other side, germ-free mice, which lack an intestinal microbiome, exhibited reduced Kyn levels, highlighting the importance of gut microbiota for the induction of IDO activity [32].
Among Trp catabolites, indoles play a pivotal role in human physiology, contributing to several functions, such as metabolic homeostasis, and immune system maturation and stimulation [33]. Some bacteria express enzymes responsible for Trp catabolism, more specifically decarboxylase and tryptophanase. Their action leads to Trp conversion into tryptamine and indole metabolites, including indole pyruvic acid, indole propionic acid (IPA), indole acetaldehyde, indole acetic acid (IAA), indole aldehyde (IAld), and indoxyl sulfate in the liver [21]. Indoles are key effectors in the gut, and they are known to be endogenous ligands of the Aryl Hydrocarbon Receptor (AHR) [21]. AHR is a transcription factor principally known to control environmental toxicity by binding exogenous xenobiotic toxic chemicals [34]. It also conveys microbiota-generated indole protective effects through antimicrobial peptide (AMP) production and mucosal protection from inflammation through IL-22 production [35][36]. Accordingly, IPA, an indole metabolite has been shown to improve intestinal barrier function through activation of the Pregnane X Receptor (PXR) [37]. Trp catabolism is therefore a key modulator of host–gut microbiota crosstalk and any perturbation impacting this mutualistic relationship can lead to disease development. For instance, a deficiency in the host gene caspase recruitment domain family member 9 (Card9), involved in the immune response against microorganisms, was shown to alter the microbiota composition and function, failing to produce indoles and contributing to intestinal inflammation [36]. Moreover, the disequilibrium between Kyn and indole production has been shown to affect metabolic and intestinal diseases, such as obesity [38] and celiac disease [39]. Increasing evidence has suggested a strong relationship between gut microbiota and its derived metabolites with CVD [40]. In this regard, patients with inflammatory bowel disease (IBD) have an increased risk of atherothrombotic complications [41], suggesting a link between the intestine and the cardiovascular system. Given the importance of Trp metabolites in gastrointestinal homeostasis, they may play a significant role in CVD. In this regard, patients with chronic kidney disease exhibit an accumulation of indoxyl sulfate due to insufficient renal removal, suggested to be involved in the occurrence of CVD in these patients through enhanced oxidative stress [42]. Moreover, indoxyl sulfate has been shown to induce vascular smooth muscle cell (SMC) proliferation that may sustain neointimal formation during uremia [43]. However, the causative relationship between indoxyl sulfate and CVD has not yet been fully proven and the plasma levels of indoxyl sulfate achieved in chronic kidney disease are supraphysiological, which may not be representative of what occurs in CVD patients without severe renal insufficiency. On the other hand, certain indole metabolites seem to exert anti-inflammatory effects [44]. Therefore, future studies are needed to investigate the contribution of intestinal Trp metabolites in CVD.
3. Indoleamine 2, 3-Dioxygenase 1
IDO is a rate limiting enzyme implicated in Trp catabolism via KP. It was discovered in rabbit small intestine by Hayaishi and colleagues in 1967 [45][46]. IDO is a monomeric protein of ~45 kDa constituted of 403 AAs and encoded by Ido-1 gene located in a pericentromeric region on chromosome 8.p12-p11 in humans and 8 A2 in mice. It is a widely expressed non-secreted cytosolic enzyme [47]. When produced, it exists in an inactive, heme-free, apoenzyme form. It becomes an active holoenzyme after the binding of heme, superoxide anion, and its substrate L-Trp [48].
Trp catabolism was originally proposed to be an innate immune defense mechanism to protect the host against bacterial or viral infection. Initial studies conducted by Pfefferkon et al. showed that interferon-γ (IFN-γ)-induced Trp degradation by human fibroblasts was responsible for blocking the intracellular parasite Toxoplasma gondii growth in these cells [49]. It was also considered as a central mediator of immune tolerance in pregnancy and a regulator of adaptive immune response [50]. On the same note, macrophage Trp catabolism was shown to inhibit T cell proliferation [51]. Later, it was shown that these immunoregulatory effects rely on the depletion of Trp in the microenvironment and/or the generation of biologically active metabolites.
Data from literature show a complex role of tissue-specific IDO activity involved in the generation of Trp metabolites, which could have either deleterious or protective effects in different pathological settings. Moreover, the discrepancy of the observed roles of IDO could be potentially explained by tissue-specific effects of this enzyme, suggesting that its function is tailored to the tissue and the environmental cues.
In recent years, IDO has been implicated in the pathophysiology of several inflammatory diseases, including infection, allergy, autoimmunity, chronic inflammation, inflammatory neurologic diseases, transplantation, and cancer [52]. The involvement of IDO in such diverse diseases is due to its large spectrum of expression in different organs and tissues, such as the spleen, brain, intestine, and lung [47].
During inflammation, IDO is up-regulated mostly in macrophages and dendritic cells by pro-inflammatory stimuli, such as IFN-γ [53]. IDO is an upstream enzyme which is involved in the generation of Kyn, and thereafter, due to different downstream other enzymes, the generation of Kyn-derived metabolites that have been described to exert either protective or deleterious effects [54][55]. For example, Quina plays a direct role in the pathogenesis of neurodegenerative disorders through the activation of N-methyl-d-aspartate (NMDA) receptors [54][56]. In contrast, Kna is neuroprotective through its antagonism on NMDA receptors [57].
As a protective role, IDO has been described as immunosuppressive enzyme that suppresses effector T Helper (TH17) -cell function and favors the differentiation of regulatory T cells (Tregs) [52]. Some KP-derived metabolites, particularly Kyn and Kna, were identified as endogenous ligands of AHR [58][59]. As such, Kyn was shown to cross the T-cell membrane by the system L-type amino acid transporter SLC7A5 before it activates the AHR by binding to its hydrophobic ligand-binding pocket [60]. Recently, two trace-active derivatives of Kyn named trace-extended aromatic condensation products (TEACOPs) have been identified and considered as high affinity AHR ligands [61]. Moreover, IDO expression was shown to be maintained due to an IDO-Kyn/AHR-IDO feed-forward loop [62]. IDO-mediated effects also rely on Trp consumption. Notably, Trp depletion induces the stress-response enzyme, general control nondepressible 2 (GCN2) kinase, which inhibits the anabolic mammalian target of rapamycin (mTOR) [63]. GCN2 activation in T cells can inhibit their proliferation and promote T cell differentiation toward Treg cells. GCN2 can also directly affect the phenotype of dendritic cells and macrophages by inducing an anti-inflammatory response [64].
Independently of its enzymatic activity, IDO can act as a signal transducer conferring a tolerogenic phenotype to plasmacytoid dendritic cells (pDCs) in transforming growth factor (TGF)-β-dependent manner [65]. This regulatory role of IDO may be highly relevant in the context of acquired peripheral tolerance, e.g., in pregnancy, graft tolerance, cancer, chronic infection, autoimmunity, and allergic inflammation [66].
In addition, the biological effects of IDO may go beyond its role in the regulation of the immune response, suggesting a more complex and prominent role than previously thought. IDO activity was shown to contribute to arterial vessel relaxation and to the control of blood pressure in septic shock [67]. Moreover, metabolites generated from Kyn may regulate diverse cellular functions, including viability [68], adhesive, and migratory properties [69], as well as inflammatory potential. As such, Trp metabolism has been involved in various diseases, ranging from chronic granulomatous disease [70] to neurodegenerative diseases, such as Alzheimer’s or Huntington’s disease [54]. Despite the progress made to understand the modes of action of IDO, more studies are warranted to comprehend the profile of IDO expression and activation as well as its precise role in immune-dependent diseases.
References
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25.
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82.
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction (2018). J. Am. Coll. Cardiol. 2018, 72, 2231–2264.
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 119–177.
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126.
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. New Engl. J. Med. 2005, 352, 1685–1695.
- Libby, P.; Ridker, P.M.; Hansson, G.K. Inflammation in Atherosclerosis: From Pathophysiology to Practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505.
- Zhenyukh, O.; González-Amor, M.; Díez, R.R.; Esteban, V.; Ruiz-Ortega, M.; Salaices, M.; Mas, S.; Briones, A.M.; Egido, J. Branched-chain amino acids promote endothelial dysfunction through increased reactive oxygen species generation and inflammation. J. Cell. Mol. Med. 2018, 22, 4948–4962.
- Nitz, K.; Lacy, M.; Atzler, D. Amino Acids and Their Metabolism in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 319–330.
- Eussen, S.J.; Ueland, P.M.; Vollset, S.E.; Nygård, O.; Midttun, Ø.; Sulo, G.; Ulvik, A.; Meyer, K.; Pedersen, E.R.; Tell, G.S. Kynurenines as predictors of acute coronary events in the Hordaland Health Study. Int. J. Cardiol. 2015, 189, 18–24.
- Pedersen, E.R.; Svingen, G.F.T.; Schartum-Hansen, H.; Ueland, P.M.; Ebbing, M.; Nordrehaug, J.E.; Igland, J.; Seifert, R.; Nilsen, R.M.; Nygård, O. Urinary excretion of kynurenine and tryptophan, cardiovascular events, and mortality after elective coronary angiography. Eur. Hear. J. 2013, 34, 2689–2696.
- Pedersen, E.R.; Tuseth, N.; Eussen, S.J.; Ueland, P.M.; Strand, E.; Svingen, G.F.T.; Midttun, Ø.; Meyer, K.; Mellgren, G.; Ulvik, A.; et al. Associations of Plasma Kynurenines With Risk of Acute Myocardial Infarction in Patients with Stable Angina Pectoris. Arter. Thromb. Vasc. Biol. 2015, 35, 455–462.
- Pedersen, E.R.; Midttun, Ø.; Ueland, P.M.; Schartum-Hansen, H.; Seifert, R.; Igland, J.; Nordrehaug, J.E.; Ebbing, M.; Svingen, G.; Bleie, Ø.; et al. Systemic Markers of Interferon-γ–Mediated Immune Activation and Long-Term Prognosis in Patients With Stable Coronary Artery Disease. Arter. Thromb. Vasc. Biol. 2011, 31, 698–704.
- Saadi, M.; Vodovar, N.; Paven, E.; Sadoune, M.; Barnabas, G.; Desrumeaux, G.; Sirol, M.; Mercadier, J.-J.; Launay, J.; Logeart, D. Plasma Indoleamine 2,3-dioxygenase is predictive of left ventricular remodeling after myocardial infarction. Arch. Cardiovasc. Dis. Suppl. 2020, 12, 144.
- Hopkins, F.G.; Cole, S.W. A contribution to the chemistry of proteids. J. Physiol. 1901, 27, 418–428.
- Richard, D.M.; Dawes, M.A.; Mathias, C.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. 2009, 2, IJTR–S2129–60.
- Palego, L.; Betti, L.; Rossi, A.; Giannaccini, G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J. Amino Acids 2016, 2016, 1–13.
- Keszthelyi, D.; Troost, F.; Masclee, A.A.M. Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol. Motil. 2009, 21, 1239–1249.
- Taleb, S. Tryptophan Dietary Impacts Gut Barrier and Metabolic Diseases. Front. Immunol. 2019, 10, 2113.
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938.
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. 2019, 10, 249.
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., III; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2014, 29, 1395–1403.
- Zhang, X.; Beaulieu, J.-M.; Sotnikova, T.D.; Gainetdinov, R.; Caron, M.G. Tryptophan Hydroxylase-2 Controls Brain Serotonin Synthesis. Science 2004, 305, 217.
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276.
- Walther, D.J.; Peter, J.U.; Bashammakh, S.; Hortnagl, H.; Voits, M.; Fink, H.; Bader, M. Synthesis of Serotonin by a Second Tryptophan Hydroxylase Isoform. Science 2003, 299, 76.
- Kulikova, E.A.; Kulikov, A.V. Tryptophan hydroxylase 2 as a therapeutic target for psychiatric disorders: Focus on animal models. Expert Opin. Ther. Targets 2019, 23, 655–667.
- Borghi, M.; Puccetti, M.; Pariano, M.; Renga, G.; Stincardini, C.; Ricci, M.; Giovagnoli, S.; Costantini, C.; Romani, L. Tryptophan as a Central Hub for Host/Microbial Symbiosis. Int. J. Tryptophan Res. 2020, 13, 1178646920919755.
- Choi, S.-C.; Brown, J.; Gong, M.; Ge, Y.; Zadeh, M.; Li, W.; Croker, B.P.; Michailidis, G.; Garrett, T.J.; Mohamadzadeh, M.; et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci. Transl. Med. 2020, 12, eaax2220.
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703.
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.J.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673.
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724.
- Zhang, N. The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J. Cardiovasc. Dis. Res. 2011, 2, 91–95.
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385.
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; Costa, G.D.A.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605.
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.; et al. Symbiotic Bacterial Metabolites Regulate Gastrointestinal Barrier Function via the Xenobiotic Sensor PXR and Toll-like Receptor 4. Immunity 2014, 41, 296–310.
- Laurans, L.; Venteclef, N.; Haddad, Y.; Chajadine, M.; Alzaid, F.; Metghalchi, S.; Sovran, B.; Denis, R.; Dairou, J.; Cardellini, M.; et al. Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health. Nat. Med. 2018, 24, 1113–1120.
- Lamas, B.; Hernandez-Galan, L.; Galipeau, H.J.; Constante, M.; Clarizio, A.; Jury, J.; Breyner, N.M.; Caminero, A.; Rueda, G.; Hayes, C.L.; et al. Aryl hydrocarbon receptor ligand production by the gut microbiota is decreased in celiac disease leading to intestinal inflammation. Sci. Transl. Med. 2020, 12, eaba0624.
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 1–17.
- Cainzos-Achirica, M.; Glassner, K.; Zawahir, H.S.; Dey, A.K.; Agrawal, T.; Quigley, E.M.; Abraham, B.P.; Acquah, I.; Yahya, T.; Mehta, N.N.; et al. Inflammatory Bowel Disease and Atherosclerotic Cardiovascular Disease. J. Am. Coll. Cardiol. 2020, 76, 2895–2905.
- Konopelski, P.; Ufnal, M. Indoles—Gut Bacteria Metabolites of Tryptophan with Pharmacotherapeutic Potential. Curr. Drug Metab. 2018, 19, 883–890.
- Chen, W.-J.; Lai, Y.-J.; Lee, J.-L.; Wu, S.-T.; Hsu, Y.-J. CREB/ATF3 signaling mediates indoxyl sulfate-induced vascular smooth muscle cell proliferation and neointimal formation in uremia. Atherosclerosis 2020, 315, 43–54.
- Wlodarska, M.; Luo, C.; Kolde, R.; D’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.e6.
- Higuchi, K.; Hayaishi, O. Enzymic formation of d-kynurenine from d-tryptophan. Arch. Biochem. Biophys. 1967, 120, 397–403.
- Yamamoto, S.; Hayaishi, O. Tryptophan pyrrolase of rabbit intestine. D- and L-tryptophan-cleaving enzyme or enzymes. J. Biol. Chem. 1967, 242, 5260–5266.
- Dai, X.; Zhu, B.T. Indoleamine 2,3-Dioxygenase Tissue Distribution and Cellular Localization in Mice: Implications for Its Biological Functions. J. Histochem. Cytochem. 2009, 58, 17–28.
- Yeung, A.W.; Terentis, A.; King, N.J.C.; Thomas, S.R. Role of indoleamine 2,3-dioxygenase in health and disease. Clin. Sci. 2015, 129, 601–672.
- Pfefferkorn, E.R. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA 1984, 81, 908–912.
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of Allogeneic Fetal Rejection by Tryptophan Catabolism. Science 1998, 281, 1191–1193.
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1372.
- Mellor, A.L.; Munn, D.H. Ido expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774.
- Chon, S.Y.; Hassanain, H.H.; Gupta, S.L. Cooperative Role of Interferon Regulatory Factor 1 and p91 (STAT1) Response Elements in Interferon-γ-inducible Expression of Human Indoleamine 2,3-Dioxygenase Gene. J. Biol. Chem. 1996, 271, 17247–17252.
- Schwarcz, R.; Köhler, C. Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci. Lett. 1983, 38, 85–90.
- Gheorghe, C.E.; Martin, J.; Manriquez, F.V.; Dinan, T.G.; Cryan, J.F.; Clarke, G. Focus on the essentials: Tryptophan metabolism and the microbiome-gut-brain axis. Curr. Opin. Pharmacol. 2019, 48, 137–145.
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of quinolinic acid on human astrocytes morphology and functions: Implications in Alzheimer’s disease. J. Neuroinflammation 2009, 6, 36.
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 2011, 145, 863–874.
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203.
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic Acid Is a Potent Endogenous Aryl Hydrocarbon Receptor Ligand that Synergistically Induces Interleukin-6 in the Presence of Inflammatory Signaling. Toxicol. Sci. 2010, 115, 89–97.
- Sinclair, L.V.; Neyens, D.; Ramsay, G.; Taylor, P.M.; Cantrell, R.A. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat. Commun. 2018, 9, 1981.
- Seok, S.-H.; Ma, Z.-X.; Feltenberger, J.B.; Chen, H.; Chen, H.; Scarlett, C.; Lin, Z.; Satyshur, K.; Cortopassi, M.; Jefcoate, C.R.; et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J. Biol. Chem. 2018, 293, 1994–2005.
- Li, Q.; Harden, J.L.; Anderson, C.D.; Egilmez, N.K.; Anderson, C. Tolerogenic Phenotype of IFN-γ–Induced IDO+ Dendritic Cells Is Maintained via an Autocrine IDO–Kynurenine/AhR–IDO Loop. J. Immunol. 2016, 197, 962–970.
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143.
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207.
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878.
- Lob, S.; Königsrainer, A. Role of IDO in Organ Transplantation: Promises and Difficulties. Int. Rev. Immunol. 2009, 28, 185–206.
- Wang, Y.; Liu, H.; McKenzie, G.; Witting, P.K.; Stasch, J.-P.; Hahn, M.; Changsirivathanathamrong, D.; Wu, B.; Ball, H.; Thomas, S.R.; et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat. Med. 2010, 16, 279–285.
- Löb, S.; Königsrainer, A.; Rammensee, H.-G.; Opelz, G.; Terness, P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: Can we see the wood for the trees? Nat. Rev. Cancer 2009, 9, 445–452.
- Barth, M.C.; Ahluwalia, N.; Anderson, T.; Hardy, G.J.; Sinha, S.; Alvarez-Cardona, J.A.; Pruitt, I.E.; Rhee, E.P.; Colvin, R.A.; Gerszten, R.E. Kynurenic Acid Triggers Firm Arrest of Leukocytes to Vascular Endothelium under Flow Conditions. J. Biol. Chem. 2009, 284, 19189–19195.
- Romani, L.; Fallarino, F.; DE Luca, A.; Montagnoli, C.; D’Angelo, C.; Zelante, T.; Vacca, C.; Bistoni, F.; Fioretti, M.C.; Grohmann, U.; et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nat. Cell Biol. 2008, 451, 211–215.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
2 times
(View History)
Update Date:
13 Oct 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No