 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | CHANGWON KHO | + 3068 word(s) | 3068 | 2021-10-10 08:18:05 | | | |
| 2 | Conner Chen | Meta information modification | 3068 | 2021-10-15 09:22:57 | | |
Video Upload Options
In hearts, calcium (Ca2+) signaling is a crucial regulatory mechanism of muscle contraction and electrical signals that determine heart rhythm and control cell growth. Ca2+ signals must be tightly controlled for a healthy heart, and the impairment of Ca2+ handling proteins is a key hallmark of heart disease. The discovery of microRNA (miRNAs) as a new class of gene regulators has greatly expanded our understanding of the controlling module of cardiac Ca2+ cycling. Furthermore, many studies have explored the involvement of miRNAs in heart diseases.
1. Introduction
Ca2+ is an important signaling molecule in all cell types and regulates the fundamental functions of various organs. The heart uses Ca2+ to maintain the cardiac rhythm and muscle function. Ca2+-induced signals also cause cardiac cell damage or death due to hypoxia. The heart pumps blood throughout the body as a key organ in the circulatory system. The adult human heart is composed of many types of cells that combine to form complex structures. Cardiac muscle cells or cardiomyocytes occupy ~75% of the structural volume [1] and are responsible for permanent blood flow by producing contractile force in intact hearts. Approximately three billion cardiomyocytes are activated by both electrical and mechanical stimuli to contract simultaneously.
As the typical ratio of the Ca2+ concentration to cytosolic concentration in the external cellular environment is close to 1:20,000, tight control of Ca2+ access to cells and efficient means of pumping Ca2+ are required. Changes in its concentration are responsible for many metabolic processes in the human body. For each beat, the intracellular free Ca2+ concentration in cardiomyocytes ([Ca2+]i) increases above 1 µM, allowing interaction between the contractile elements during systole and diastole, and [Ca2+]i decreases to approximately 100 nM, causing dissociation of the contractile elements [2]. It induces relaxation, allowing the heart to refill the blood. To elevate [Ca2+]i, extracellular Ca2+ enters the cytoplasm across the plasma membrane (sarcolemma), or intracellular Ca2+ is released from Ca2+ storage organelles, including the sarcoplasmic reticulum (SR). In cardiomyocytes, the sarcolemma Na+/Ca2+ exchanger (NCX) is the primary mechanism by which Ca2+ is released from cells. Activation of the sarco/endoplasmic reticulum Ca2+-adenosine triphosphatase 2 (SERCA2) pump promotes the reuptake of cytosolic Ca2+ into the SR. Numerous other molecules are involved in Ca2+ homeostasis in cardiomyocytes [3]. Cellular extrinsic and intrinsic signals influence cardiac function by modulating the magnitude and timing of Ca2+ transients in various cardiac regulating pathways. A summary of Ca2+ cycling observed in cardiomyocytes is presented in Figure 1.
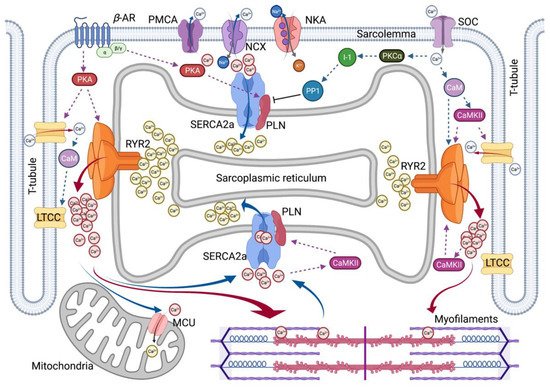
Figure 1. Overview of Intracellular Ca2+ Cycling in Cardiomyocytes. Schematic overview summarizes excitation–contraction coupling in cardiomyocytes. βAR, β–adrenergic receptor; CaM, calmodulin; CaMKII, Ca2+/CaM-dependent kinase II; I-1, Inhibitor-1; LTCC, L-type Ca2+ channel; MCU, Mitochondrial Ca2+ uniporter; NCX, Na+/Ca2+ exchanger; NKA, Na⁺/K⁺-ATPase; PKA, Protein kinase A; PKCα, Protein kinase C α-isoform; PLN, Phospholamban; PMCA, Plasma membrane Ca2+–ATPase; PP1, Protein phosphatase-1; RYR2, Ryanodine receptor type-2; SERCA2a, Sarco/endoplasmic reticulum Ca2+-adenosine triphosphatase 2; and SOC, store-operated channel. Red solid arrow represents Ca2+-induced Ca2+ release. Blue solid arrow represents Ca2+ extrusion in diastolic relaxation. Dotted lines represent a positive or negative effect on the following molecules.
2. Excitation–Contraction Coupling
The contraction movement of hearts is carried out by excitation–contraction coupling (ECC), which occurs in cardiomyocytes through Ca2+ regulation [3]. Ca2+ regulation in cardiomyocytes is maintained by the action of specific proteins, such as Ca2+ channels, pumps, transporters, and exchangers. The SR serves as an important repository and sinks with the regulation of [Ca2+]i during ECC, and in cardiomyocytes, most Ca2+ is stored in the SR within the milli-molar range. Ca2+-buffering proteins (e.g., calsequestrin, CSQ) within the SR lumen are important Ca2+ sensors and contribute to deciphering Ca2+ transients and signals during ECC.
Cardiac ECC is the process whereby an electrical stimulation (i.e., excitation) of the surface membrane with an action potential (AP) triggers a cardiomyocyte to depolarize and contract. Ca2+ plays two pivotal roles in the ECC. Ca2+ drives myofilament activation and regulates sarcolemma ionic currents that are responsible for normal electrical rhythms. A series of ECC events in cardiomyocytes are as follows: (1) initiation and propagation of an AP along with the plasma membrane, (2) rapid spread of the potential along with the transverse tubule system (T-tubule system), (3) dihydropyridine receptors (DHPR, L-type Ca2+ channel CaV1.1, LTCC)-mediated detection of changes in membrane potential, (4) ryanodine receptors (RyRs) stimulation due to allosteric interaction of the LTCC with the SR RyRs, (5) release of Ca2+ through stimulated RyRs in SR and transient increase in [Ca2+]i, (6) transient activation of the cytosolic Ca2+ buffering system and the contractile apparatus, followed by (7) disappearance of Ca2+ from the cytosol mediated by its movement to the mitochondria via the mitochondrial Ca2+ uniporter (MCU), its expulsion by the NCX and plasma membrane Ca2+–ATPase (PMCA) at the sarcolemma, and its final reuptake by the SR through the SERCA2. Both the duration and intensity of cardiac AP affect the regulation of Ca2+ fluxes and contractility in cardiomyocytes.
In addition, ECC is regulated by multiple signaling pathways. The β–adrenergic pathway is regulated by β-agonists, such as adrenaline, which activate the β–adrenergic receptor (βAR) and initiates the production of cyclic adenosine monophosphate (cAMP) by adenylate cyclase, which activates protein kinase A (PKA). Another pathway is the Ca2+-mediated calmodulin (CaM)-dependent kinase (CaMK) signaling, which is activated by increased cytosolic Ca2+content, thereby inducing regulation of ECC. More recent studies have reported a process called excitation–transcription coupling (ETC), which reveals a similar role of Ca2+ in controlling gene transcription in cardiomyocytes to that of Ca2+-dependent signaling of excitation–contraction coupling [4].
3. Two Major Players in SR Calcium Flux in Heart Disease
ECC defects that cause contractile dysfunction and cardiac arrhythmia are typical features of HF. In failing cardiomyocytes, a major defect in Ca2+ cycling occurs at the SR. Abnormal expression and function of key Ca2+ handling proteins, including SERCA2a, RyR2, phospholamban (PLN), CaM, CSQ2, triadin, and junctin, result in excessive [Ca2+]i, decreased SR Ca2+ uptake, increased SR Ca2+ leak, and decreased SR Ca2+ content. SERCA2a and RyR2 are primarily responsible for the sequestration and release of SR Ca2+. Both SERCA2a and RyR2 are regulated by post-translational modifications (PTMs) and interacting partner proteins. Therefore, strategies to restore impaired SR Ca2+ homeostasis caused by an abnormality in these two Ca2+ handling proteins have been extensively studied for the treatment of HF. The major SR Ca2+-handling proteins reported to be associated with HD are summarized in Table 1.
Table 1. SR Calcium Handling Protein Associated with Human Heart Diseases.
| Protein | Disease Phenotype | References |
|---|---|---|
| RyR2 | CPVT, ARVD/C2, AF | Priori et al., 2001 [5]; Marks et al., 2002 [6]; Yano et al., 2005 [7] |
| SERCA2a | HF | Hasenfuss et al., 1994 [8]; Meyer et al., 2006 [9]; Flesch et al., 1996 [10] |
| PLN | ARVD, DCM | Zwaag et al., 2012 [11]; Jordan et al., 2021 [12] |
| CSQ2 | CPVT | Lahat et al., 2001 [13]; Postma et al., 2002 [14] |
| CaM | CPVT, LQTS | Chazin and Johnson. 2020 [15] |
| Triadin | CPVT | Roux-Buisson et al., 2012 [16]; Rooryck et al., 2015 [17] |
| Junctin | DCM | Gergs et al. 2007 [18] |
| HRC | Arrhythmias, DCM, AF | Arvanitis et al., 2008 [19]; Amioka et al., 2019 [20] |
AF, atrial fibrillation; ARVD, arrhythmogenic right ventricular dysplasia; ARVD/C2 arrhythogenic right ventricular cardiomyopathy type 2; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; HF, heart failure; and LQTS, long QT syndrome.
3.1. SERCA2a Calcium Pump
One of the most striking cellular changes in failing human hearts is an increase in end-diastolic [Ca2+]i and prolongation of diastolic Ca2+ decay. SERCA2a is the dominant SERCA isoform in hearts and determines the clearance of more than 70% of cytosolic Ca2+ in humans and 90% of cytosolic Ca2+ in rodents [3]. Dysregulation of SERCA2a is a hallmark of HF. Several studies have shown that SERCA activity is diminished in failing human and animal hearts. Indeed, normalization of SERCA2a expression has been shown to significantly improve contractility and Ca2+ homeostasis in failing human cardiomyocytes [21] and increase hemodynamics with antiarrhythmic effects in rodent and large animal models of HF [22][23]. Therefore, restoration of SERCA2a function is an attractive therapeutic approach. SERCA2a gene therapy, which increases gene expression, and small molecule drugs, which stimulate enzyme activity, are being developed as new treatments for chronic HF. Several clinical studies have been conducted to correct SERCA2a enzyme abnormalities, such as CUPID (Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease) 1, CUPID 2, AGENT-HF (AAV1-CMV-Serca2a Gene Therapy Trial in Heart Failure), and SERCA-LVAD trial [24][25][26][27]. These studies showed that SERCA2a gene delivery is safe and has potential benefits in advanced HF.
The typical underlying mechanism for reduced SERCA2a activity is inhibition by interacting with partner proteins. PLN is a reversible regulator of SERCA2a. PLN binds directly to SERCA2a and inhibits its affinity for Ca2+. The inhibition of SERCA2a by PLN is regulated by the phosphorylation of PLN rather than by changes in PLN expression. It has been observed that PLN phosphorylation is reduced in the heart tissues of most patients with HF [28][29]. βAR stimulation leads to PLN phosphorylation at the serine-16 site by PKA and threonine-17 site by CaMKII or protein kinase B (AKT). Phosphorylated PLN loses its binding to SERCA2a, which in turn increases SERCA2a pump activity. In addition, several PLN-binding proteins, such as hematopoietic lineage cell-specific protein-1 associated protein X-1 (HAX-1), intra-luminal histidine-rich Ca2+ binding protein (HRC), S100A1, and protein phosphatase 1 (PP1), contribute to PLN-dependent SERCA2a regulation [30]. In 2016, Nelson et al. discovered a muscle-specific long noncoding RNA called the dwarf open reading frame (DWORF) as a novel activator of SERCA. The DWORF peptide has been proposed to indirectly activate SERCA2a by displacing PLN [31]. However, more diverse studies are needed to determine the exact physiological role of DWORF in the heart. Some studies have reported a reduction in PLN mRNA levels in patients with dilated or ischemic cardiomyopathy [10]. Notably, pathogenic variants in PLN, known to be associated with hereditary dilated cardiomyopathy (DCM) with HF, have been reported to cause problems with binding affinity for SERCA2a. Genetic correction of PLN mutations via genome editing techniques combined with gene transfer produced positive results, including normalized Ca2+ handling in a patient-derived cell model of DCM, suggesting a novel strategy for DCM treatment [32]. In particular, with strong support from the Leducq Foundation, an international network of excellence program in cardiovascular research, PLN-induced cardiomyopathy studies have become intensive [33].
SERCA2a function is also regulated by PTMs, including nitrosylation, glutathionylation, glycation, SUMOylation, and acetylation. In particular, SUMO1 deficiency and decreased SUMOylation levels of SERCA2a have been observed in failing hearts [34]. Restoration of SUMO1 via gene transfer has explored the therapeutic potential of targeting SUMOylation as a method to increase SERCA2a activity and improve cardiac contractility in both mouse and porcine models of HF [34][35][36]. Furthermore, this PTM framework provides a new perspective on SERCA2a modulation, resulting in the identification of novel SUMO-SERCA2a activators [37]. Recent work has uncovered additional roles of deacetylation/acetylation in modulating SERCA2a function during HF [38]. Overall, these PTMs are considered a fine-tuning mechanism of SERCA2a, which may enhance the capacity of active targeting SERCA2a to treat HF.
3.2. RyR2 Calcium Release Channel
RyR2 is a gate-keeping mechanism that serves as a Ca2+-induced Ca2+ release channel on the SR during the systole of cardiomyocytes. The amount of SR Ca2+ released by RyR2 is correlated with the strength of the systolic contraction. RyR2 is the largest ion channel in nature (four monomers of 565 kDa each). RyR2 combines with numerous accessory proteins to form a large macromolecular complex, including FKBP (FK506-binding protein 12.6), CaM, CSQ2, junctin, triadin, HRC, S100A1, and Sorcin [39]. They control the Ca2+ sensitivity of RyR2 through dynamic interactions, which in turn alters the probability of RyR2 opening or closing. For example, loss or dysfunction of CSQ2, the main SR Ca2+-binding protein, exposes RyR2 to excess free SR Ca2+. As another example, defective CaM binding to RyR2 destabilizes the channel. Indeed, several CaM mutations are associated with severe forms of long QT syndrome (LQTS) and catecholaminergic polymorphic ventricular tachycardia (CPVT) [15].
In addition, the gating ability of RyR2 is regulated by various PTMs, including phosphorylation, oxidation, and nitrosylation [40]. Many studies have shown that SR Ca2+ leaks are caused by increased sensitivity of RyRs to Ca2+ due to RyR phosphorylation by kinases (i.e., PKA or CaMKII) or phosphatases (i.e., phosphatase 1 or 2A). In particular, phosphorylation of RyR2 to specific serine residues, such as Ser-2808, Ser-2814, and Ser-2030, appears to induce functional changes in RyR2, and an association with HD has been reported [41]. For example, PKA-mediated RyR2 induces the dissociation of FKBP, a RyR2 stabilizer, which increases the probability of opening. In this context, prolonged PKA phosphorylation may lead to premature contractions and arrhythmias. However, contradictory results have been reported for RyR2-mediated Ca2+ spark via phosphorylation in intact cardiomyocytes [41][42].
Under pathogenic conditions such as HF, RyR2-mediated SR Ca2+ release continues during diastole, reducing SR Ca2+ content. RyR2 leakage can be due to altered expression levels of RyR2-associated proteins or dysregulation of PTMs, such as enhanced PKA- and CaMKII-dependent phosphorylation and reduced phosphatase activity. The exacerbating effects of SR Ca2+ leakage on cardiac function include: (1) decreased systolic SR Ca2+ levels leading to systolic dysfunction, (2) elevation of diastolic Ca2+ leading to diastolic dysfunction, (3) energy outflow to re-pump Ca2+, and (4) induced arrhythmias. Although there is no change in the protein expression of RyR2 in patients with HF, abnormal Ca2+ sparks are observed in failing cardiomyocytes [43]. The exact mechanisms and regimes of SR operation that generate abnormal Ca2+ leaks remain elusive. Several abnormal functions of RyR2 have been identified in patients with CPVT, arrhythmic right ventricular cardiomyopathy (ARVC), or atrial filtration (AF) as the causes of disease-related mutations and interacting partners [5][6][7][44].
Regarding therapeutics, RyR2 gene replacement approaches are limited owing to the vector size. Alternatively, siRNA delivery to silence mutant mRNA of RyR2 in an allele-specific manner or genome-editing approach has proven effective in normalizing cardiac electrophysiology in a mouse model of CPVT [45]. Several types of drugs have been developed that target mutated or dysfunctional RyR2 channels, including benzothiazepine derivatives (K201 and S107), tetracaine derivatives (EL9 and EL20), and unnatural verticilide enantiomers [46]. The potential of K201 for the treatment of AF has been evaluated clinically; however, the results have not been reported. Several FDA-approved drugs modulate RyR2, including flecainide and propafenone, which are anti-arrhythmic agents that act as Na+ channel blockers with additional activity in the open state of RyR2. Dantrolene, a pan-RyR inhibitor, has been investigated for its anti-arrhythmic efficacy in patients with CPVT (NCT04134845) [46].
4. Calcium Signaling through Protein Kinases
As described above, Ca2+ cycling is achieved through harmony with vital Ca2+-handling proteins strictly regulated by changes in PTMs, such as phosphorylation. The most relevant modulator for these proteins is PKA, which can directly phosphorylate and regulate major proteins in cardiomyocytes, including LTCC, RyR2, and PLN. A further mechanism of control is provided by CaMKII, targeting the same Ca2+ handling proteins.
4.1. β–Adrenergic Receptor-Mediated PAK Regulation
PKA is a cAMP-dependent protein kinase that has multiple roles in the regulation of cardiac function, including contraction, metabolism, ion flux, and gene transcription. A principle underlying the mechanism of cardiac β–adrenergic receptor (βAR) signal transduction is as follows: in response to stress, the binding of agonist βARs selectively interacts with the stimulatory G protein to directly stimulate adenylyl cyclase, converting ATP to cAMP, which activates PKA. βAR-mediated PKA activation phosphorylates several ECC proteins such as LTCC, RyR, cardiac troponin I (cTnI), cardiac myosin binding protein C (cMyBPC), and PLN. An additional critical target in Ca2+ cycling is inhibitor-1 (I-1), which is also controlled by PKA. I-1 becomes active upon PKA phosphorylation and inhibits type 1 serine/threonine protein phosphatase (PP1), resulting in amplification of βAR responses in hearts.
Chronic HF is associated with increased sympathetic nervous system activity [47]. In the early stages of reduced cardiac function, an increase in sympathetic activity preserves cardiac output. As heart function deteriorates, activation of neurohumoral signaling pathways increases to compensate for disease progression. However, prolonged neurohormonal activation causes significant damage to cardiomyocytes. Under long-term stimulation, βAR signaling is regulated by the coordinated action of at least three enzymes, both at the receptor level and downstream of the cascade: G protein-coupled receptor kinases, which phosphorylate and desensitize the receptor; cyclic nucleotide phosphodiesterases, which degrade cAMP; and phosphatases, which dephosphorylate phosphoproteins. Chronic stimulation of βAR results in multiple changes in the ARAR signaling cascade, including downregulation of βAR, upregulation of βAR kinase, and increased inhibitory G-protein α-subunit function. In this context, it is a foregone conclusion that abnormalities in the βAR-PKA pathway are important determinants of cardiac dysfunction and HF. Reduced PKA activity and decreased phosphorylation of downstream targets such as PLN, cTnI, and cMyBP, have been observed in patients with AF, hypertrophic cardiomyopathy, and HF [48][49][50]. However, increased protein levels and activity of PKA have also been reported in failing hearts [51]. PKA-mediated hyperphosphorylation of RyR2 is found in dysfunctional human and canine hearts, associated with RyR2 channel instability creating leaky channels. Although there are contradictory reports on the expression level and activity of PKA in diseased hearts, aberrant PKA activation or inactivation contributes to the pathogenesis of myocardial ischemia, hypertrophy, and HF.
4.2. Calcium-Calmodulin Mediated CaMKII Regulation
CaMKII is another critical regulatory kinase responsible for the phosphorylation of key ECC proteins and the transcriptional activation of pathological hypertrophy [52][53]. CaMKII is inactive in normal status, but is activated by increased [Ca2+]i and reactive oxygen species (ROS). Initially, the elevation of [Ca2+]i triggers Ca2+binding to CaM, activating CaMKII. Activation of CaMKII by Ca2+-CaM depends on the local Ca2+ level and the frequency of Ca2+ release. When [Ca2+]i increases briefly, CaMKII returns to its inactive form after Ca2+-CaM dissociation. However, the continuous presence of Ca2+-CaM allows for autophosphorylation of CaMKII, which causes “CaM trapping” to maintain CaMKII activity even at low [Ca2+]i conditions. For example, CaMKII can be activated through PTMs induced by ROS independent of Ca2+-CaM but also activated in response to βAR/PKA stimulation [54].
CaMKII is considered a major pathogenic signaling molecule in HD [55][56][57]. For example, upregulation of CaMKII activity and expression appears to be a common hallmark of cardiomyopathy of various etiologies in patients and animal models, suggesting that CaMKII is a signaling molecule in cardiomyopathy. Given the vital role of CaMKII in ion channel regulation, CaMKII seems to behave as a pro-arrhythmogenic protein in the heart. The expression and activity of CaMKII increase in AF, resulting in the promotion of arrhythmogenesis. Besides arrhythmias, oxidized, constitutively active CaMKII has been strongly linked with ischemia/reperfusion injury (I/R), diabetes cardiomyopathy, and HF.
References
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575.
- Fearnley, C.J.; Roderick, H.L.; Bootman, M.D. Calcium Signaling in Cardiac Myocytes. Cold Spring Harb. Perspect. Biol. 2011, 3, a004242.
- Bers, D.M. Cardiac excitation—Contraction coupling. Nature 2002, 415, 198–205.
- Dewenter, M.; von der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020.
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Danieli Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200.
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194.
- Dridi, H.; Kushnir, A.; Zalk, R.; Yuan, Q.; Melville, Z.; Marks, A.R. Intracellular calcium leak in heart failure and atrial fibrillation: A unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 2020, 17, 732–747.
- Hasenfuss, G.; Reinecke, H.; Studer, R.; Meyer, M.; Pieske, B.; Holtz, J.; Holubarsch, C.; Posival, H.; Just, H.; Drexler, H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994, 75, 434–442.
- Meyer, M.; Schillinger, W.; Pieske, B.; Holubarsch, C.; Heilmann, C.; Posival, H.; Kuwajima, G.; Mikoshiba, K.; Just, H.; Hasenfuss, G. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 1995, 92, 778–784.
- Flesch, M.; Schwinger, R.H.; Schnabel, P.; Schiffer, F.; van Gelder, I.; Bavendiek, U.; Südkamp, M.; Kuhn-Regnier, F.; Böhm, M. Sarcoplasmic reticulum Ca2+ATPase and phospholamban mRNA and protein levels in end-stage heart failure due to ischemic or dilated cardiomyopathy. J. Mol. Med. 1996, 74, 321–332.
- Van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 199–207.
- Hershberger, R.E.; Jordan, E. Dilated Cardiomyopathy Overview. GeneReviews . 2007. Available online: https://pubmed.ncbi.nlm.nih.gov/20301486/ (accessed on 27 September 2021).
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384.
- Postma, A.V.; Denjoy, I.; Hoorntje, T.M.; Lupoglazoff, J.M.; da Costa, A.; Sebillon, P.; Mannens, M.M.; Wilde, A.A.; Guicheney, P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2002, 91, e21–e26.
- Chazin, W.J.; Johnson, C.N. Calmodulin Mutations Associated with Heart Arrhythmia: A Status Report. Int. J. Mol. Sci. 2020, 21, 1418.
- Roux-Buisson, N.; Cacheux, M.; Fourest-Lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767.
- Rooryck, C.; Kyndt, F.; Bozon, D.; Roux-Buisson, N.; Sacher, F.; Probst, V.; Thambo, J.B. New Family With Catecholaminergic Polymorphic Ventricular Tachycardia Linked to the Triadin Gene. J. Cardiovasc. Electrophysiol. 2015, 26, 1146–1150.
- Gergs, U.; Berndt, T.; Buskase, J.; Jones, L.R.; Kirchhefer, U.; Müller, F.U.; Schlüter, K.D.; Schmitz, W.; Neumann, J. On the role of junctin in cardiac Ca2+ handling, contractility, and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H728–H734.
- Arvanitis, D.A.; Sanoudou, D.; Kolokathis, F.; Vafiadaki, E.; Papalouka, V.; Kontrogianni-Konstantopoulos, A.; Theodorakis, G.N.; Paraskevaidis, I.A.; Adamopoulos, S.; Dorn, G.W., 2nd; et al. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur. Heart J. 2008, 29, 2514–2525.
- Amioka, M.; Nakano, Y.; Ochi, H.; Onohara, Y.; Sairaku, A.; Tokuyama, T.; Motoda, C.; Matsumura, H.; Tomomori, S.; Hironobe, N.; et al. Ser96Ala genetic variant of the human histidine-rich calcium-binding protein is a genetic predictor of recurrence after catheter ablation in patients with paroxysmal atrial fibrillation. PLoS ONE 2019, 14, e0213208.
- Del Monte, F.; Harding, S.E.; Schmidt, U.; Matsui, T.; Kang, Z.B.; Dec, G.W.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999, 100, 2308–2311.
- Lyon, A.R.; Bannister, M.L.; Collins, T.; Pearce, E.; Sepehripour, A.H.; Dubb, S.S.; Garcia, E.; O’Gara, P.; Liang, L.; Kohlbrenner, E.; et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 2011, 4, 362–372.
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119.
- Hajjar, R.J.; Zsebo, K.; Deckelbaum, L.; Thompson, C.; Rudy, J.; Yaroshinsky, A.; Ly, H.; Kawase, Y.; Wagner, K.; Borow, K.; et al. Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J. Card. Fail. 2008, 14, 355–367.
- Jaski, B.E.; Jessup, M.L.; Mancini, D.M.; Cappola, T.P.; Pauly, D.F.; Greenberg, B.; Borow, K.; Dittrich, H.; Zsebo, K.M.; Hajjar, R.J. Calcium Up-Regulation by Percutaneous Administration of Gene Therapy In Cardiac Disease (CUPID) Trial Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail. 2009, 15, 171–181.
- Hulot, J.S.; Salem, J.E.; Redheuil, A.; Collet, J.P.; Varnous, S.; Jourdain, P.; Logeart, D.; Gandjbakhch, E.; Bernard, C.; Hatem, S.N.; et al. AGENT-HF Investigators. Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: Results from the AGENT-HF randomized phase 2 trial. Eur. J. Heart Fail. 2017, 19, 1534–1541.
- Lyon, A.R.; Babalis, D.; Morley-Smith, A.C.; Hedger, M.; Suarez Barrientos, A.; Foldes, G.; Couch, L.S.; Chowdhury, R.A.; Tzortzis, K.N.; Peters, N.S.; et al. Investigation of the safety and feasibility of AAV1/SERCA2a gene transfer in patients with chronic heart failure supported with a left ventricular assist device—The SERCA-LVAD TRIAL. Gene Ther. 2020, 27, 579–590.
- Schmidt, U.; Hajjar, R.J.; Kim, C.S.; Lebeche, D.; Doye, A.A.; Gwathmey, J.K. Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. Am. J. Physiol. 1999, 277, H474–H480.
- Schwinger, R.H.; Munch, G.; Bolck, B.; Karczewski, P.; Krause, E.G.; Erdmann, E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J. Mol. Cell. Cardiol. 1999, 31, 479–491.
- Haghighi, K.; Bidwell, P.; Kranias, E.G. Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J. Mol. Cell. Cardiol. 2014, 77, 160–167.
- Nelson, B.R.; Makarewich, C.A.; Anderson, D.M.; Winders, B.R.; Troupes, C.D.; Wu, F.; Reese, A.L.; McAnally, J.R.; Chen, X.; Kavalali, E.T.; et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 2016, 351, 271–275.
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955.
- Doevendans, P.A.; Glijnis, P.C.; Kranias, E.G. Leducq Transatlantic Network of Excellence to Cure Phospholamban-Induced Cardiomyopathy (CURE-PLaN). Circ. Res. 2019, 125, 720–724.
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605.
- Lee, A.; Jeong, D.; Mitsuyama, S.; Oh, J.G.; Liang, L.; Ikeda, Y.; Sadoshima, J.; Hajjar, R.J.; Kho, C. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid. Redox Signal. 2014, 21, 1986–2001.
- Tilemann, L.; Lee, A.; Ishikawa, K.; Aguero, J.; Rapti, K.; Santos-Gallego, C.; Kohlbrenner, E.; Fish, K.M.; Kho, C.; Hajjar, R.J. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci. Transl. Med. 2013, 5, 211ra159.
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; deVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229.
- Gorski, P.A.; Jang, S.P.; Jeong, D.; Lee, A.; Lee, P.; Oh, J.G.; Chepurko, V.; Yang, D.K.; Kwak, T.H.; Eom, S.H.; et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca2+-ATPase in Heart Failure. Circ. Res. 2019, 124, e63–e80.
- Zalk, R.; Lehnart, S.E.; Marks, A.R. Modulation of the ryanodine receptor and intracellular calcium. Annu. Rev. Biochem. 2007, 76, 367–385.
- Federico, M.; Valverde, C.A.; Mattiazzi, A.; Palomeque, J. Unbalance Between Sarcoplasmic Reticulum Ca2+ Uptake and Release: A First Step Toward Ca2+ Triggered Arrhythmias and Cardiac Damage. Front. Physiol. 2020, 10, 1630.
- Dobrev, D.; Wehrens, X.H. Role of RyR2 Phosphorylation in Heart Failure and Arrhythmias. Circ. Res. 2014, 114, 1311–1319.
- Li, Y.; Kranias, E.G.; Mignery, G.A.; Bers, D.M. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ. Res. 2002, 90, 309–316.
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376.
- Belevych, A.E.; Radwanski, P.B.; Carnes, C.A.; Gyorke, S. ‘Ryanopathy’: Causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc. Res. 2013, 98, 240–247.
- Bongianino, R.; Denegri, M.; Mazzanti, A.; Lodola, F.; Vollero, A.; Boncompagni, S.; Fasciano, S.; Rizzo, G.; Mangione, D.; Barbaro, S.; et al. Allele-Specific Silencing of Mutant mRNA Rescues Ultrastructural and Arrhythmic Phenotype in Mice Carriers of the R4496C Mutation in the Ryanodine Receptor Gene (RYR2). Circ. Res. 2017, 121, 525–536.
- Patrick, C.; Tarah, A.W.; Xander, H.T.W. Targeting Pathological Leak of Ryanodine Receptors: Preclinical Progress and the Potential Impact on Treatments for Cardiac Arrhythmias and Heart Failure. Expert Opin. Ther. Targets 2020, 24, 25–36.
- Packer, M. The neurohormonal hypothesis: A theory to explain the mechanism of disease progression in heart failure. J. Am. Coll. Cardiol. 1992, 20, 248–254.
- El-Armouche, A.; Boknik, P.; Eschenhagen, T.; Carrier, L.; Knaut, M.; Ravens, U.; Dobrev, D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 2006, 114, 670–680.
- Zakhary, D.R.; Moravec, C.S.; Stewart, R.W.; Bond, M. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation 1999, 99, 505–510.
- Jacques, A.M.; Copeland, O.; Messer, A.E.; Gallon, C.E.; King, K.; McKenna, W.J.; Tsang, V.T.; Marston, S.B. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J. Mol. Cell. Cardiol. 2008, 45, 209–216.
- Wang, J.; Liu, X.; Arneja, A.S.; Dhalla, N.S. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can. J. Cardiol. 1999, 15, 683–690.
- Maier, L.S.; Zhang, T.; Chen, L.; DeSantiago, J.; Brown, J.H.; Bers, D.M. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 2003, 92, 904–911.
- Zhang, M.; Hagenmueller, M.; Riffel, J.H.; Kreusser, M.M.; Bernhold, E.; Fan, J.; Katus, H.A.; Backs, J.; Hardt, S.E. Calcium/calmodulin-dependent protein kinase II couples Wnt signaling with histone deacetylase 4 and mediates dishevelled-induced cardiomyopathy. Hypertension 2015, 65, 335–344.
- Erickson, J.R. Mechanisms of CaMKII activation in the heart. Front. Pharmacol. 2014, 5, 59.
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. CaMKII: Linking heart failure and arrhythmias. Circ. Res. 2012, 110, 1661–1677.
- Bell, J.R.; Vila-Petroff, M.; Delbridge, L.M. CaMKII-dependent responses to ischemia and reperfusion challenges in the heart. Front. Pharmacol. 2014, 5, 96.
- Hegyi, B.; Bers, D.M.; Bossuyt, J. CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 127, 246–259.


