 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hannah Pook | + 2327 word(s) | 2327 | 2021-10-08 12:06:11 | | | |
| 2 | Beatrix Zheng | + 575 word(s) | 2902 | 2021-10-11 05:26:33 | | | | |
| 3 | Hannah Pook | Meta information modification | 2902 | 2021-10-11 14:44:18 | | |
Video Upload Options
Pancreatic ductal adenocarcinoma (PDAC) is a type of cancer that arises in the exocrine glands of the pancreas and comprises over 90% of pancreatic malignancies. Currently the 11th most common cancer worldwide, PDAC is the seventh leading cause of cancer-related deaths and is on track to move to second place by 2030. Despite the high prevalence, therapeutic options remain limited, with only modest improvements in overall survival (OS) occurring over the past 50 years.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a type of cancer that arises in the exocrine glands of the pancreas [1] and comprises over 90% of pancreatic malignancies [2]. Currently the 11th most common cancer worldwide [3], PDAC is the seventh leading cause of cancer-related deaths and is on track to move to second place by 2030 [1]. Despite the high prevalence, therapeutic options remain limited, with only modest improvements in overall survival (OS) occurring over the past 50 years [4]. The 5-year overall survival is currently just 8% [5], with median survival time remaining under 1 year even with newer treatments [6], highlighting a clear need for new and effective therapies. Despite extensive research in the area, surgery is still the only curative option, although late presentation and high rates of metastases by the time of detection limit this approach to just 20% of newly diagnosed cases [7]. Even in those suitable for surgery, re-emergence occurs in 90% following the operation [8]. The current outlook for patients with PDAC continues to be poor and highlights a clear need for new therapeutic options.
Cytotoxic chemotherapy remains the mainstay of treatment in PDAC [9], although research has led to optimised combinations to provide improvements in OS. The PRODIGE-24 trial placed modified 5-fluoro-uracil (5-FU), leucovorin, irinotecan, and oxaliplatin (mFOLFIRINOX) as the standard-of-care adjuvant therapy following significantly increased OS in comparison to gemcitabine (54.4 months vs. 35 months) [10], while in patients with advanced-stage disease, combinations such as paclitaxel and gemcitabine or FOLFIRINOX have also elicited modestly improved OS [6][11]. Research aiming to generate new therapies has had a broad focus, likely accounted for by the highly heterogenous nature of PDAC cancers and lack of actionable genetic aberrations. The most common mutation, which occurs in the KRAS proto-oncogene and is seen in >95% of human PDACs [12], is unfortunately largely unactionable. This may be set to change, however, due to the emergence of direct KRAS inhibitors able to target the G12C mutant KRAS variant [13][14]. Although this mutation is present in just 1–4% of PDACs [15], and hence the majority of patients will be unable to benefit from this inhibitor if it becomes clinically available, its development is a major step forward in targeting a protein previously thought to be undruggable.
Other strategies currently being investigated include pathway inhibition, metabolic targeting, DNA damage, immunotherapeutic agents, epigenetic targeting, tumour microenvironment (TME) modification, and destruction of cancer stem cells (CSCs). Pre-clinical successes have been seen in many of these fields, although, as of yet, they have not translated into clinical practice. Outside of therapeutics themselves, the determination of biomarkers to guide drug selection and improvements in pancreatic cancer screening and detection are areas that will be key in improving patient outcomes.
It is now well established within the field that PDACs can be stratified into different molecular subtypes, which impact OS and therapeutic response [16][17]. This was first noted by Collison in 2011 [18], who defined three subtypes (classical, quasi-mesenchymal, and exocrine like) using gene expression microarray analysis in primary resected samples as well as human and mouse cell lines. Several years later, Moffitt et al. [19] used virtual microdissection to digitally separate tumour and stromal gene expression to specify two tumour-specific and two stromal-specific subtypes, while Bailey et al. [20] carried out an integrated genomic analysis to determine four distinct subtypes, as summarised in Table 1. More recent research has served to validate the existence of such subtypes and highlight the significant overlap between these classifications, indicating similarities between classical and pancreatic progenitor subtypes as well as basal-like and squamous tumours [16]. Given the molecular differences arising between subtypes, for example, elevated GATA6 expression in the classical/pancreatic progenitor subtype [18][19][21], they could prove to be a useful modality for patient stratification to determine personalised medicine regimes and thus improve patient outcomes. However, further work still needs to be carried out to unify current classification methods and produce a single comprehensive grouping strategy. This would facilitate the application of knowledge to both clinical and research purposes, making precision medicine more viable in the context of PDAC going forwards.
| Classification | Subtype | Characteristics |
|---|---|---|
| Collison | Classical | Better prognosis than QM subtype following resection High expression of adhesion-associated and epithelial genes, high GATA6 expression Increased dependence on KRAS and increased sensitivity to erlotinib compared to QM subtype |
| Quasi-mesenchymal (QM) | High expression of mesenchyme associated genes Increased sensitivity to gemcitabine compared to classical subtype |
|
| Exocrine-like | Relatively high expression of tumour cell-derived digestive enzyme genes | |
| Moffitt tumour-specific subtypes | Classical | 20/22 genes shared with Collisson classical subtype |
| Basal-like | Worse median survival and 1-year survival compared to basal-like subtype Better response to adjuvant therapy compared to classical subtype High expression of laminins and keratins |
|
| Moffitt stromal-specific subtypes | Normal | High expression of markers for pancreatic stellate cells, smooth muscle actin, vimentin, and desmin |
| Activated | Worse median and 1-year survival compared to normal subtype More diverse gene expression including genes associated with macrophages (e.g., ITGAM, CCL13, and CCL18) and genes involved in tumour promotion (e.g., SPARC, WNT2, WNT5A, MMP9) |
|
| Bailey | Squamous | Poor prognosis compared to other subtypes High expression of TP53 and KDM6A, and upregulation of TP63∆N transcriptional network Hypermethylation of pancreatic endodermal cell-fate determining genes |
| Pancreatic progenitor | High expression of genes involved in early pancreatic development (FOXA2/3, PDX1, and MNX1) | |
| Immunogenic | Significant immune infiltrate Upregulation of immune networks, including pathways associated with acquired immune suppression |
|
| Aberrantly differentiated endocrine exocrine | Subclass of pancreatic progenitor tumours High expression of genes involved in regulating KRAS activation, exocrine (NR5A2 and RBPJL), and endocrine differentiation (NEUROD1 and NKX2-2) |
2. Immunotherapies
Despite being widely regarded as an immunologically ‘cold’ cancer type there has been increasing interest in the potential of immunotherapies to treat PDAC ( Figure 1 ). Disappointingly, single-checkpoint blockade therapies have been shown to be ineffective in treating patients [22][23], with resistance to this form of immunotherapy being multifactorial in nature. A lower mutational burden compared to malignancies known to respond to immune checkpoint inhibitors (ICIs) [24], an immunosuppressive microenvironment, and a dense desmoplastic stroma that act as a physical barrier to immune cell infiltration are all thought to be contributing factors [25]. However, it is possible to modulate these factors to increase immune-responsiveness and this has proven to be an attractive approach in recent research.
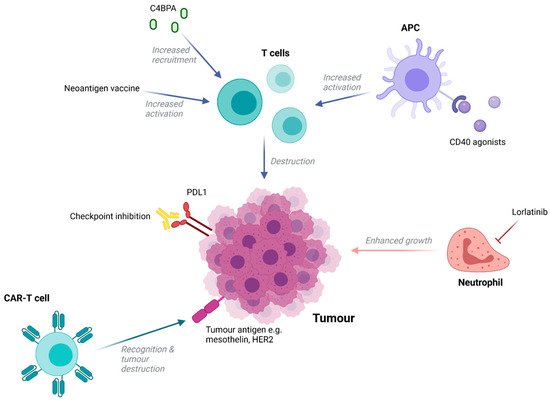
For example, agonists to CD40, a member of the TNF receptor superfamily commonly expressed in immune cells, enabled antigen-presenting cells to increase levels of T-cell activation in tumour explants from a mouse KPC model [26]. A phase I trial in which an agonistic anti-CD40 antibody was administered in combination with nab-paclitaxel and gemcitabine with or without nivolumab demonstrated a 58% response rate amongst 24 patients [27], highlighting the potential of this approach. Alternatively, recent studies have shown that complement performs seminal functions in promoting or suppressing cancer progression. New research indicated that high levels of stromal C4b binding protein alpha chain (C4BPA) within human PDAC tissue were associated with better prognosis, including longer OS, and strongly correlated with increased levels of CD8+ tumour-infiltrating lymphocytes [28]. In vivo, GnP/ICBs/mC4BPA peptide treatment, but not GnP treatment, led to the accumulation of a greater number of CD8+ T cells in the periphery of PDAC tumours and increased tumour regression [28]. This highlights C4BPA stimulation as another potential method that could convert PDAC to an immunologically ‘hot’ tumour and thus increase the curative potential of ICIs.
Outside of ICIs, multiple immunotherapies have been trialled with varying results. Mesothelin-targeting CAR-T cells have been trialled in six patients and were well tolerated, with one patient experiencing a 69% decrease in metabolically active tumour volume [29]. However, as with other solid tumours, lack of accessibility for circulating CAR-T cells and a rarity of uniformly expressed targetable epitopes is likely to hinder therapeutic efficacy. Nonetheless, additional trials involving CAR-T cells targeting mesothelin and other antigens are ongoing. Additionally, novel work using a specific subset of effector memory T-cells expressing CD161 as opposed to bulk T cells to generate HER2-specific CARs demonstrated the potential to improve CAR-T constructs in PDAC [30]. Importantly, the use of this particular T-cell population was able to enhance cytotoxicity and improve tumour burden control in mouse models of pancreatic cancer when compared to bulk peripheral mononuclear blood cells.
New research has also aimed to target immune cell types outside of T cells. Neutrophils are abundant within the PDAC TME and are associated with poor clinical prognoses. They are known to promote tumour growth through ECM modulation and cytokine production and are also thought to aid in metastasis by supporting the survival of circulating cancer cells, enhancing stem cell characteristics of metastasis initiating cells, and establishing a hospitable niche for metastasis growth [31][32][33]. This highlights neutrophils as a promising target, with their inhibition being able to prevent both cancer growth and spread. Lorlatinib, a small molecular tyrosine kinase inhibitor able to suppress neutrophil accumulation, development, and activity, was able to attenuate PDAC progression and growth at both primary and metastatic sites in a PDAC mouse model [34]. Furthermore, when combined with an anti-PD-1 blockade, lorlatinib increased the levels of activated intra-tumoural CD8+ T cells and resulted in significantly smaller tumours compared to control and monotherapy groups [34].
3. Extracellular Tumour Microenvironment Modification
The abundance of fibrotic stroma, known as desmoplasia and derived mainly from pancreatic stellate cells, is a feature typical of PDAC. This dense extracellular matrix acts as a physical barrier preventing delivery of therapeutic agents to tumours and barring entry of immune cells to contribute to the ‘immunologically cold’ TME [35][36][37]. Hence, approaches that target the tumour stroma, in addition, to directly targeting PDAC cells are attractive. One strategy involved targeting focal adhesion kinase (FAK), a tyrosine kinase that promotes tumour invasion, growth, and metastasis through interactions with stromal cells in a number of solid malignancies [38]. In a KPC mouse model of PDAC, FAK inhibition reduced fibrosis and improved survival [36], with clinical trials investigating combined FAK inhibition and ICIs or cytotoxic agents now underway. Furthermore, positive results have been observed upon targeting connective tissue growth factor (CTGF), which is highly expressed in preclinical models of PDAC and is thought to contribute to desmoplasia [39]. Pamrevlumab, an anti-CTGF monoclonal antibody, led to improved OS in PDAC patients in combination with standard chemotherapy in a phase I/II trial [40]. On the basis of these results, pamrevlumab has now been designated as a fast-track therapy to treat patients with locally advanced PDAC. However, there is evidence that suggests that components of the stroma may act to restrain cancer cell growth and impede metastasis [41], with reports that poorly differentiated PDAC with low desmoplasia is more aggressive and has a worse prognosis [42]. Thus, caution must be taken when targeting the stroma in order to ensure that the effects are beneficial.
This is illustrated in the case of targeting hyaluronic acid (HA), a glycosaminoglycan (GAG) overexpressed in PDAC stroma and associated with low immune response and poor prognosis [43][44]. HA was initially thought to be a promising target in PDAC, with one study involving HA staining on samples from 101 patients with stage IA-IIB disease revealing that high levels of stroma HA expression were significantly associated with poor disease-specific survival and OS [45]. In mouse models, treatment with PEGPH20 (a pegylated recombinant human hyaluronidase that breaks down hyaluronan) and gemcitabine prolonged survival [46], while a phase II trial administering PEGPH20 and nab-paclitaxel/gemcitabine to patients with advanced-stage PDAC found that the addition of PEGPH20 significantly improved PFS in patients with high levels of hyaluronic acid expression [47]. However, despite these early encouraging results, a phase III trial (HALO-301) involving the administration of PEGPH20 plus nab-paclitaxel/gemcitabine (PAG) or placebo plus nab-paclitaxel/gemcitabine (AG) failed to meet its primary endpoints [48]. Disappointingly, OS in the PAG group was slightly lower than the AG group (11.2 months vs. 11.7 months) and there was no difference in PFS between the two. Furthermore, results from a phase Ib/II trial in which PEGPH20 was combined with mFOLFIRINOX in patients with metastatic PDAC demonstrated that PEGPH20 addition was detrimental to OS [49], re-iterating the potential adverse consequences of stromal targeting. Alternatively, trial failure could have resulted from the targeting of desmoplasia alone being insufficient to evoke treatment benefits with other factors such as low tumour mutational burden, lack of targetable neoantigens, and EMT also requiring modulation to produce favourable results [50].
4. Elimination of Cancer Stem Cells
Cancer stem cells (CSCs) refer to a specific subset of cells within tumours, which display self-renewal and the ability to produce differentiated progeny, characteristics typically associated with normal stem cells. First described in 1997 in acute myeloid leukaemia [51], CSCs have since been discovered in multiple other cancer types, with the first report of their presence in PDAC arising in 2007 [52]. Since then, pancreatic CSCs (PCSCs) have been shown to play a vital role in PDAC propagation and growth [53]. More recently, they have been linked to tumour stroma differentiation, with a greater presence of PCSC markers being associated with a loose stroma type and higher rates of cumulative local recurrence [54]. Furthermore, PCSCs are known to contribute to chemoresistance in PDAC [55] and cancer recurrence following treatment, hence curing PDAC is contingent on complete CSC annihilation.
Much work has been carried out in this area aiming to characterise and selectively target pancreatic CSCs. JAK-STAT and Hedgehog pathways, known to be involved in CSC maintenance [56][57], have been targeted in PDAC but failed to produce satisfactory results [58], highlighting a need for better characterisation of CSC signalling. Alternatively, it is possible to target specific surface markers of CSCs, which include CD44, CD24, CD133, CXCR4, and ESA [52][53]. For example, the generation of a bi-specific antibody recognising ESA and CD3 was able to re-direct cytotoxic T lymphocytes to eliminate highly tumourigenic PCSCs in vitro and in vivo [59].
Specific metabolic targeting of CSCs is also a promising approach, with PCSCs highlighted as being dependent on oxidative phosphorylation in (OXPHOS) in order to survive. Inhibition of this pathway by metformin was able to produce rapid apoptosis specifically within the PDAC CSC population while non-CSC cells were less dramatically affected, only undergoing cell cycle arrest [60]. Furthermore, recent work on four PDAC cell lines has shown that PCSCs have specific and common proteome and lipidome modulations, which could prove to be useful therapeutic targets [61]. These include mitochondrial cardiolipin acyl chain modification and upregulated fatty acid elongation and phosphoinositol phosphatase pathways, features unique to PCSCs and possible targets for drug development.
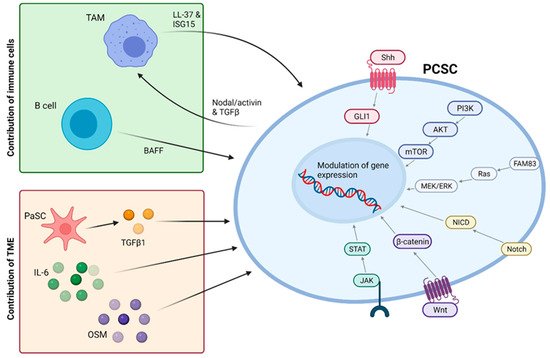
However, it is important to note that increasing bodies of work suggest that PCSCs, and CSCs as a whole, are not a defined subpopulation but rather a state that non-CSC tumour cells are also able to enter [62]. This means that not only is it important to target existing CSCs but also to understand and inhibit pathways promoting cellular plasticity that may give rise to new CSCs. Such pathways have been extensively reviewed by Smigiel et al. (2018) [58] and are summarised in Figure 2 . Many of these targets have already demonstrated promising pre-clinical results, with combined targeting of mTOR and c-MET signalling eliciting a dramatic reduction in the viability of CD133+ CSCs while the inhibition of Notch signalling by genistein significantly reduced self-renewal of PCSCs [58]. Furthermore, pharmacological inhibition or knockdown of ALK4, the target receptor of Nodal and Activin, demonstrated the ability to abolish the self-renewal and tumourigenicity of PCSCs and sensitised this subpopulation to gemcitabine [63].
References
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385.
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 1–22.
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27.
- Roser, M.; Ritchie, H. Cancer. Our World in Data. Available online: https://ourworldindata.org/cancer (accessed on 6 August 2021).
- Hill, A.; Chung, V. Pancreatic Cancer. Oncol. Precis. Med. Era 2020, 97–109.
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825.
- Li, J.; Wientjes, M.G.; Au, J.L.-S. Pancreatic Cancer: Pathobiology, Treatment Options, and Drug Delivery. AAPS J. 2010, 12, 223–232.
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy with Gemcitabine Plus Erlotinib versus Gemcitabine Alone in Patients after R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337.
- NICE. Pancreatic Cancer in Adults: Diagnosis and Management. NICE Guideline . Available online: https://www.nice.org.uk/guidance/ng85/chapter/Recommendations#ftn.footnote_5 (accessed on 8 August 2021).
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406.
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703.
- Dreyer, S.; Chang, D.K.; Bailey, P.; Biankin, A.V. Pancreatic Cancer Genomes: Implications for Clinical Management and Therapeutic Development. Clin. Cancer Res. 2017, 23, 1638–1646.
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17.
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003.
- Waters, A.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435.
- Aguirre, A.J. Refining Classification of Pancreatic Cancer Subtypes to Improve Clinical Care. Gastroenterology 2018, 155, 1689–1691.
- Martens, S.; Lefesvre, P.; Nicolle, R.; Biankin, A.; Puleo, F.; Van Laethem, J.; Rooman, I. Different shades of pancreatic ductal adenocarcinoma, different paths towards precision therapeutic applications. Ann. Oncol. 2019, 30, 1428–1436.
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503.
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178.
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52.
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13.
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti–PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293.
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833.
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421.
- Upadhrasta, S.; Zheng, L. Strategies in Developing Immunotherapy for Pancreatic Cancer: Recognizing and Correcting Multiple Immune “Defects” in the Tumor Microenvironment. J. Clin. Med. 2019, 8, 1472.
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6Clow F4/80+ Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210.
- O’Hara, M.H.; O’Reilly, E.M.; Rosemarie, M.; Varadhachary, G.; Wainberg, Z.A.; Ko, A.; Fisher, G.A.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. Abstract CT004: A Phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine (Gem) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in untreated metastatic ductal pancreatic adenocarcinoma (PDAC) patients. Cancer Res. 2019, 79, CT004.
- Sasaki, K.; Takano, S.; Tomizawa, S.; Miyahara, Y.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Takada, M.; Ohtsuka, M. C4b-binding protein α-chain enhances antitumor immunity by facilitating the accumulation of tumor-infiltrating lymphocytes in the tumor microenvironment in pancreatic cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–16.
- Beatty, G.L.; O’Hara, M.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32.
- Konduri, V.; Joseph, S.K.; Byrd, T.T.; Nawas, Z.; Vazquez-Perez, J.; Hofferek, C.J.; Halpert, M.M.; Liu, D.; Liang, Z.; Baig, Y.; et al. A subset of cytotoxic effector memory T cells enhances CAR T cell efficacy in a model of pancreatic ductal adenocarcinoma. Sci. Transl. Med. 2021, 13, eabc3196.
- Lianyuan, T.; Gang, L.; Ming, T.; Dianrong, X.; Chunhui, Y.; Zhaolai, M.; Bin, J. Tumor associated neutrophils promote the metastasis of pancreatic ductal adenocarcinoma. Cancer Biol. Ther. 2020, 21, 937–945.
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.-S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560.
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845.
- Nielsen, S.R.; Strøbech, J.E.; Horton, E.R.; Jackstadt, R.; Laitala, A.; Bravo, M.C.; Maltese, G.; Jensen, A.R.D.; Reuten, R.; Rafaeva, M.; et al. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade. Nat. Commun. 2021, 12, 1–15.
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072.
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.; Nywening, T.M.; Hawkins, T.M.N.W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860.
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, 102655.
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610.
- Bennewith, K.L.; Huang, X.; Ham, C.M.; Graves, E.E.; Erler, J.; Kambham, N.; Feazell, J.; Yang, G.P.; Koong, A.; Giaccia, A.J. The Role of Tumor Cell–Derived Connective Tissue Growth Factor (CTGF/CCN2) in Pancreatic Tumor Growth. Cancer Res. 2009, 69, 775–784.
- Picozzi, V.J.; Pishvaian, M.J.; Mody, K.; Winter, J.M.; Glaspy, J.A.; Larson, T.; Matrana, M.R.; Saikali, K.; Carney, M.; Porter, S.; et al. Effect of anti-CTGF human recombinant monoclonal antibody pamrevlumab on resectability and resection rate when combined with gemcitabine/nab-paclitaxel in phase 1/2 clinical study for the treatment of locally advanced pancreatic cancer patients. J. Clin. Oncol. 2018, 36, 4016.
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747.
- Wang, L.M.; Silva, M.A.; D’Costa, Z.; Bockelmann, R.; Soonawalla, Z.; Liu, S.; O’Neill, E.; Mukherjee, S.; McKenna, W.G.; Muschel, R.; et al. The prognostic role of desmoplastic stroma in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 4183–4194.
- Cheng, X.-B.; Sato, N.; Kohi, S.; Yamaguchi, K. Prognostic Impact of Hyaluronan and Its Regulators in Pancreatic Ductal Adenocarcinoma. PLoS ONE 2013, 8, e80765.
- Kuang, D.-M.; Wu, Y.; Chen, N.; Cheng, J.; Zhuang, S.-M.; Zheng, L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007, 110, 587–595.
- Tahkola, K.; Ahtiainen, M.; Mecklin, J.-P.; Kellokumpu, I.; Laukkarinen, J.; Tammi, M.; Tammi, R.; Väyrynen, J.P.; Böhm, J. Stromal hyaluronan accumulation is associated with low immune response and poor prognosis in pancreatic cancer. Sci. Rep. 2021, 11, 1–9.
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429.
- Hingorani, S.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366.
- Tempero, M.A.; Van Cutsem, E.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. HALO 109-301: A randomized, double-blind, placebo-controlled, phase 3 study of pegvorhyaluronidase alfa (PEGPH20) + nab-paclitaxel/gemcitabine (AG) in patients (pts) with previously untreated hyaluronan (HA)-high metastatic pancreatic ductal adenocarcinom. J. Clin. Oncol. 2020, 38, 638.
- Ramanathan, R.K.; McDonough, S.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.R.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. A phase IB/II randomized study of mFOLFIRINOX (mFFOX) + pegylated recombinant human hyaluronidase (PEGPH20) versus mFFOX alone in patients with good performance status metastatic pancreatic adenocarcinoma (mPC): SWOG S1313 (NCT #01959139). J. Clin. Oncol. 2018, 36, 208.
- Hakim, N.; Patel, R.; DeVoe, C.; Saif, M.W. Why HALO 301 Failed and Implications for Treatment of Pancreatic Cancer. Pancreas 2019, 3, e1–e4.
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737.
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037.
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323.
- Askan, G.; Sahin, I.H.; Chou, J.F.; Yavas, A.; Capanu, M.; Iacobuzio-Donahue, C.A.; Basturk, O.; O’Reilly, E.M. Pancreatic cancer stem cells may define tumor stroma characteristics and recurrence patterns in pancreatic ductal adenocarcinoma. BMC Cancer 2021, 21, 385.
- Mueller, M.; Hermann, P.C.; Witthauer, J.; Rubio–Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D’Haese, J.G.; et al. Combined Targeted Treatment to Eliminate Tumorigenic Cancer Stem Cells in Human Pancreatic Cancer. Gastroenterology 2009, 137, 1102–1113.
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Nature 2020, 5, 1–35.
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19.
- Smigiel, J.M.; Parameswaran, N.; Jackson, M.W. Targeting Pancreatic Cancer Cell Plasticity: The Latest in Therapeutics. Cancers 2018, 10, 14.
- Cioffi, M.; Dorado, J.; Baeuerle, P.A.; Heeschen, C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin. Cancer Res. 2012, 18, 465–474.
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518.
- Di Carlo, C.; Sousa, B.C.; Manfredi, M.; Brandi, J.; Dalla Pozza, E.; Marengo, E.; Palmieri, M.; Dando, I.; Wakelam, M.J.; Lopez-Clavijo, A.F.; et al. Integrated lipidomics and proteomics reveal cardiolipin alterations, upregulation of HADHA and long chain fatty acids in pancreatic cancer stem cells. Sci. Rep. 2021, 11, 1–13.
- Hermann, P.C.; Sainz, B. Pancreatic cancer stem cells: A state or an entity? Semin. Cancer Biol. 2018, 53, 223–231.
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin Signaling Drives Self-Renewal and Tumorigenicity of Pancreatic Cancer Stem Cells and Provides a Target for Combined Drug Therapy. Cell Stem Cell 2011, 9, 433–446.


