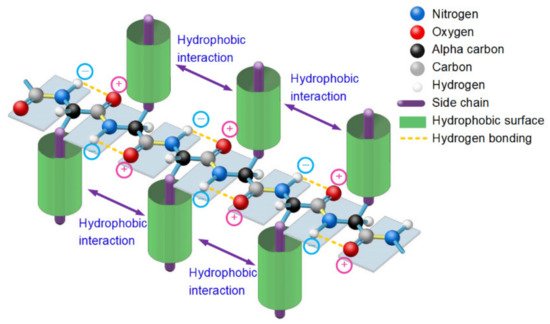
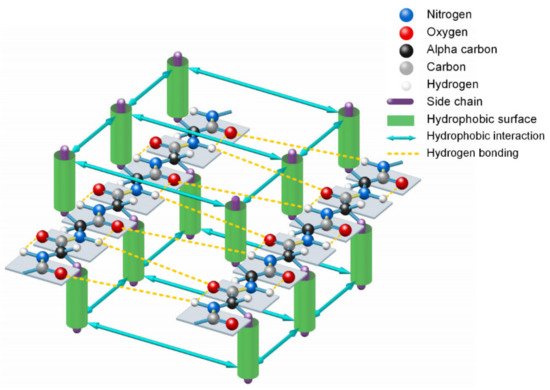
2.Entropy-Enthalpy Compensations Fold Proteins in Precise Ways
It has previously been noted that many amino acid side-chains contain considerable nonpolar sections, even if they also contain polar or charged groups
[11][12]. That is, hydrophilic side-chains are not entirely hydrophilic. The hydrophilicity of hydrophilic side-chains is normally expressed by CO or NH groups at their ends, whereas the other portions of hydrophilic side-chains are hydrophobic, because the molecular structures of these portions are basically alkyl and benzene ring structures, as shown in
Figure 1. Therefore, the folding initiation sites of secondary structures might contain not only accepted “hydrophobic” amino acids, but also long hydrophilic side-chains
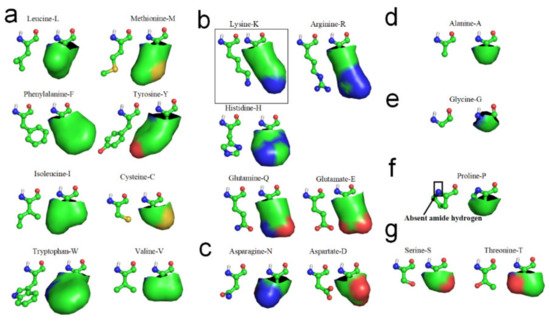
[11]. The hydrophobic portions of the hydrophilic side-chains are most likely involved in the laterally hydrophobic interaction among neighbored side-chains for secondary structures formation. Cysteine-C, Isoleucine-I, Leucine-L, Methionine-M, Tryptophan-W, Phenylalanine-F, Tyrosine-Y, and Valine-V can be fully involved in hydrophobic interaction with adjacent hydrophobic side-chains due to their high hydrophobicity (see
Figure 2a). Arginine-R, Histidine-H, Lysine-K, Glutamate-E and Glutamine-Q also can actively become involved in hydrophobic interaction with adjacent hydrophobic side-chains in sequence, due to their long hydrophilic side-chains contain long nonpolar alkyl structures, (see
Figure 2b). Aspartate-D and Asparagine-N would permit very limited participation in hydrophobic interaction with neighboring side-chains in sequence because their exposed hydrophobic proportions are relatively small (see
Figure 2c). Alanine-A most likely can laterally hydrophobic attract with long hydrophilic side-chains, due to its hydrophobic side-chain is short enough to hydrophobic attract with hydrophobic proportions of these hydrophilic side-chains and without repelling with the hydrophilic tops of these side-chains (see
Figure 2d). Glycine-G cannot effectively participate in lateral hydrophobic interaction with other neighbored side-chains in folding of a β-strand, because the hydrophobic proportion of its side-chain is negligible (see
Figure 2e). Note that Proline-P normally cannot directly contribute to the formation of β-strands through the entropy-enthalpy compensation, because Proline-P does not contain the N-H group in the main-chain (see
Figure 2f) that causes no H-bond formation between adjacent peptide planes at the residue of the backbone (see
Figure 1). Thus, Proline-P normally terminate β-strands formation. When a hydrophobic side-chain can avoid latterly approaching to the hydrophilic proportion of a hydrophilic side-chain, we can conceive that the hydrophobic side-chain can laterally hydrophobic attract the hydrophilic side-chain, as a method for predicting whether a hydrophilic side-chain can laterally hydrophobic attract another hydrophobic or hydrophilic side-chain.
Figure 1. A thermodynamically metastable state of unfolded proteins is the parallel distributed state of adjacent peptide planes due to hydrophobic interactions among neighbored side-chains and the hydrogen bonding between each carbonyl oxygen atom and adjacent amide hydrogen atom in peptide plane and the entropy-enthalpy compensation, as with a typical β-strand.
Figure 2. Hydrophobic portions of amino acid side-chains (hydrophobic portions are highlighted green). (a) Leucine, Methionine, Phenylalanine, Tyrosine, Isoleucine, Cysteine, Tryptophan, Valine. (b) Lysine, Arginine, Histidne, Glutamine, Glutamate. (c) Aspartate, Asparagine. (d) Alanine. (e) Glycine. (f) Proline. (g) Serine, Threonine.
Since the formation of β-strands is driven by hydrophobic interactions among neighboring side-chains of unfolded polypeptide in sequence and guided by the enthalpy-entropy compensation according to the Gibbs free energy equation
[13], we should be able to find experimental evidence of the hydrophobic interaction in the PDB archives. We use 1000 experimentally determined small protein structures to demonstrate and verify the hydrophobic-effect-based folding mechanism in β-sheets (see Supplementary Materials S1). All the 1000 small proteins were randomly selected from the PDB. Among them, α-type proteins accounted for 27.3%, β-type proteins accounted for 14.3%, α/β-type proteins accounted for 2.9%, and α+β-type proteins accounted for 55.5%. There are 45 similar sequences in the 1000 samples. With use of the PDB archive and the STRIDE software
[14], 3427 typical β-strands (four or more amino acids long) can be identified in the 1000 protein structures. From analysis of all the 3427 β-strands of the 1000 proteins in the PDB, we find that the phenomenon of hydrophobic side-chains or hydrophobic portions of the hydrophilic side-chains latterly clustering together (due to the hydrophobic effect) on one side or the other of β-strands is prevalent in all experimentally determined β-sheets. This finding confirmed that the hydrophobic interactions among neighboring side-chains and the entropy-enthalpy compensations are responsible for the formation of β-strands. Hydrophobic effects can contribute to the formation of β-sheets through multistage aggregations of neighboring hydrophobic groups of unfolded polypeptides and the entropy-enthalpy compensations, leading to the formation of β-strands that subsequently fold into β-sheets (see
Figure 3).
Figure 3. Lateral hydrogen bonding process of segments of two β-strands in folding a β-sheet driven by hydrophobic interactions among side-chains and entropy-enthalpy compensations.
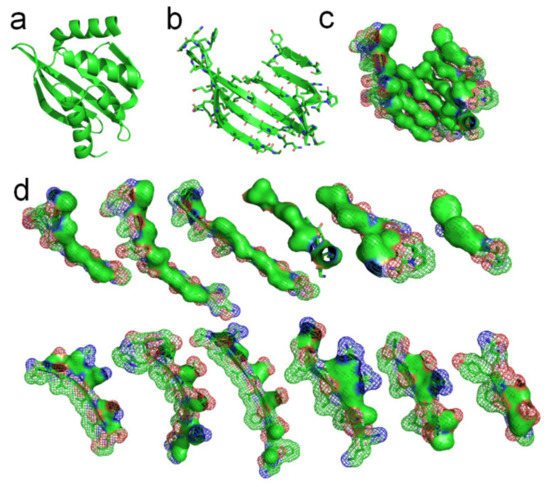
A de novo designed protein (PBDID: 5TPJ) is a good example to illustrate the phenomenon of hydrophobic attraction (due to the hydrophobic effect) among adjacent side-chains on each β-strand of a protein (see
Figure 4)
[15]. To illustrate the hydrophobic attraction, we highlight the hydrophobic surface areas of adjacent side-chains on each β-strand of the protein, based on the experimentally determined protein structure, as shown in
Figure 4c,d. Note that every β-strand is characterized by a large hydrophobic surface fully covering one side of the β-brand (the inner side), and causing each side-chain to be parallel to every other side-chain of each strand, due to the hydrophobic interaction. Parallel distribution of neighboring “hydrophobic” side-chains in a β-strand can effectively reintroduce entropy to the system via the merging of the water cages of the side-chains, which frees the ordered water molecules (see
Figure 4d). Thus, the β-strand should be considered an initial metastable state for many unfolded polypeptide segments corresponding to its free energy minimum under the solution conditions, creating localized regions of predominantly hydrophobic proportions of side-chains
[16]. Lateral hydrogen bonding process of segments of β-strands during the folding process of a β-sheet should be also driven by hydrophobic interactions among the side-chains and entropy-enthalpy compensations, as shown in
Figure 3. β-sheets folding highly depends on the temperature
[5], where β-sheets can form in as little as one microsecond after a temperature jump
[17][18][19]. The temperature dependence of folding of β-sheets is thus attributed to the temperature dependence of the Gibbs free energy equation.
Figure 4. Hydrophobic attraction among neighboring side-chains of β-strands. (a) A de novo designed protein (PBDID: 5TPJ). (b) The curved β-sheet of 5TPJ. (c) Hydrophobic attraction among adjacent β-strands via the hydrophobic surfaces of side-chains of the β-sheet (hydrophobic surfaces are highlighted green). (d) Hydrophobic surface areas on the 6 β-strands of the sheet (green areas).
The β-turn is the third most important secondary structure after helices and β-strands. Aspartate-D, Asparagine-N, Serine-S, and Glycine-G cannot effectively hydrophobic attract with neighboring side-chains in sequence because the hydrophobic proportions of their side-chains are very small (see
Figure 2). Proline-P normally cannot directly contribute to the formation of β-strands through the entropy-enthalpy compensation, since Proline-P does not contain the N-H group in the main-chain. Thus, Aspartate-D, Asparagine-N, Serine-S, Proline-P, and Glycine-G most likely lead to the formation of β-turns in protein folding, due to the tendency of the other neighboring hydrophobic side-chains in the amino acid sequence to hydrophobically collapse together by bypassing these residues. β-turns have been classified in accordance with the values of the dihedral angles φ and ψ of the central residue. β-turns can easily be identified between β-strands or α-helices of protein structures using the PDB archive and the STRIDE software
[14]. We identified 5776 β-turns in the 1000 protein structures, including about 1780 β-hairpin turns. We found that about 97.4% of the β-turns contained at least one Aspartate-D, Asparagine-N, Serine-S, Proline-P or Glycine-G residue
[20], as illustrated in
Supplementary Materials S1. Moreover, about 99.3% of β-hairpin turns contain at least one residue of Aspartate-D, Asparagine-N, Serine-S, Proline-P or Glycine-G (see
Supplementary Materials S1).
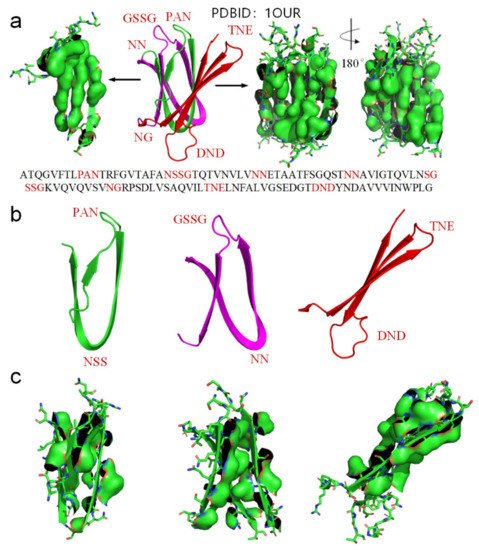
We use another small-molecule protein (PBDID:1OUR) as an example, to demonstrate the role played by hydrophobic interactions among neighboring side-chains in the formation of β-strands, β-turns, and β-sheets (see Figure 5). The protein is mainly comprised of β-strands and 10 β-turns. Every β-strand of the protein is also characterized by a large hydrophobic surface fully covering one side of the β-strand (see Figure 5a). Aspartate-D, Asparagine-N, Serine-S, Proline-P, Glycine-G contribute to the formation of β-turns in protein folding, because the other neighboring side-chains in the β-strands tend to hydrophobically attract to each other through bypassing these residues (see Figure 2). Thus, Aspartate-D, Asparagine-N, Serine-S, Proline-P, and Glycine-G can be classified as a hydrophobic blocking (RB) group. It is worth noting that almost all the 10 β-turns of the protein are composed with two or more residues of Aspartate-D, Asparagine-N, Serine-S, Proline-P, Glycine-G (see Figure 5a,b). This indicates that two or more adjacent RB residues can effectively block hydrophobic attraction among neighboring side-chains in sequence on both sides of a strand. We plot the protein structure in three parts in accordance with three segments of the amino acid sequence to illustrate the hydrophobic collapse among neighboring β-strands in sequence (see Figure 5b,c). Hydrophobic interactions among these β-strands cause them to collapse together through bending the unfolded polypeptide at the location of these RB residues. This observation also indicates that the entropy-enthalpy compensations drive hydrophobic attraction and hydrogen bonding among the β-strands to fold into the β-sheets. The formation of β-sheets also causes the β-strands to aggregate or “collapse” into a tertiary conformation with a hydrophobic core. Thereby, the folding of β-sheets is triggered by multistage hydrophobic interactions and entropy-enthalpy compensations among neighboring residues of unfolded polypeptides, enabling β-sheets to fold following explicit physical folding codes (see Figure 3, Figure 4 and Figure 5).
Figure 5. (a) Hydrophobic surface areas on the β-strands of the protein (PDBID: 1OUR), hydrophobic surface of side-chains is highlighted by green surface areas, residues located at turns are highlighted red in the protein sequence. (b) The parts of the protein (residues 1–33 highlighted green, residues 34–71 highlighted magenta, residues 72–114 highlighted red). (c) Hydrophobic surface areas on the β-strands of the sheet (green surface areas).
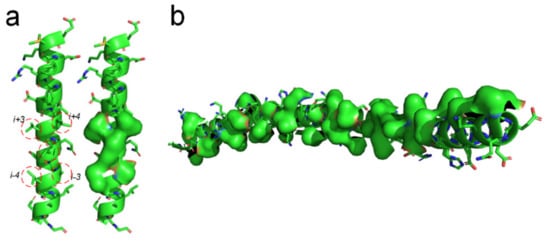
There should be entropy-enthalpy compensations that allow polypeptide chain segments to find the states of α-helices encoded in their sequence. An α-helix structure usually has a large number of hydrophobic side-chains agglomerated on its surface (see Figure 6). The folding of the α-helix structure may be also driven by the hydrophobic collapse of adjacent side-chains in the sequence through the entropy-enthalpy compensations. The typical state of a β-strand is that each residue side-chain can directly hydrophobic interact with the two adjacent residue side-chains at 1 interval in the sequence, as shown in Figure 1 and Figure 3. The side-chain of each residue in the α-helix structure can have a hydrophobic interaction with the surrounding four residue side-chains at two or three intervals in the sequence (see Figure 6a), which means that the entropy value of some polypeptide segments in forming the α-helices can be higher than that in forming the β-sheets. Therefore, the formation of the α-helix can be regarded as a further entropy-enthalpy compensation of the polypeptide segment from the β-strand-like thermodynamic metastable structure. The formation of α-helices enable laterally hydrophobic collapse among these side-chains of residues at two and three intervals in the amino acid sequence (see Figure 6). Therefore, when the amino acid sequence of a polypeptide fragment not only meets the structural requirements for β-strand, but also can have strong lateral hydrophobic interaction among the residues at three or three intervals in the sequence, it cause the polypeptide segment to fold into an α-helix instead of a β-strand. If a post-translational modification changes the critical lateral hydrophobic interactions among the residues at two or three intervals in the sequence, the polypeptide segment will most likely not fold into the α-helix due to the absence of the critical hydrophobic forces.
Figure 6. Lateral hydrophobic attraction among neighbored side-chains on α-helices. (a) Strong hydrophobic interaction among side-chains of the residues at 2 and 3 intervals in the amino acid sequence of a α-helix (PBDID: 5YM7); (b) A long α-helix with a long hydrophobic surface area on it caused by the hydrophobic side-chain distribution (PBDID: 2BEZ).
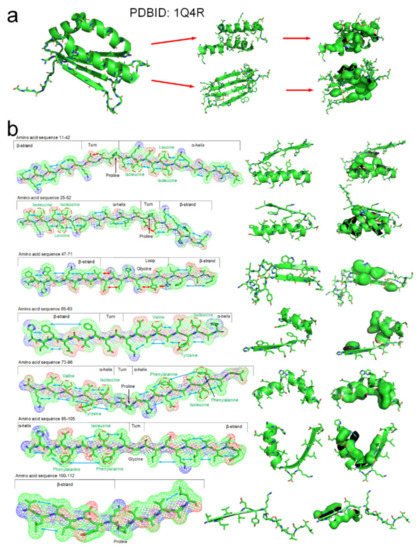
The tertiary structure of an arabidopsis protein (PDBID: 1Q4R) is a composed of typical secondary structures and is suitable as a simple example to illustrate how the entropy-enthalpy compensation mechanism can be used to predict the secondary and tertiary structures. We summarized the basic laws of laterally hydrophobic attraction and hydrophobic repulsion between side-chains of different residues. The rules of hydrophobic interaction among the side-chains of adjacent residues in the polypeptide chain sequence that causes the folding of α-helix and β-sheet are initially explored. When a fragment of a polypeptide chain in the β-strand-like thermodynamically metastable state shows sufficient hydrophobic attraction between the side-chains of adjacent residues on one side, it can be predicted that the fragment will fold into a β-strand or an α-helix. When the fragment also satisfies that a strong hydrophobic attraction can occur among the residues at two and three intervals in the sequence, it can be predicted that the polypeptide fragment will fold into an α-helix instead of a β-strand. The entropy-enthalpy compensation analysis of the amino acid sequence fragment of the protein 1Q4R is illustrated in Figure 7.
Figure 7. The folding mechanism of a protein structure (PBDID: 1Q4R) based on entropy-enthalpy compensation. (a) Hydrophobic interaction among side-chains of secondary structures. (b) The polypeptide chain fragment and the corresponding secondary structure in a thermodynamically metastable state are drawn in 7 segments (the hydrophobic attraction between the side-chains of adjacent residues is marked with a blue arrow, and the hydrophilic-hydrophobic repulsion is marked with a red arrow). The proline and glycine that led to the formation of the corner structure are marked. The hydrophobic amino acids in the sequence that cause the metastable collapse to form an α-helix structure are annotated by red circles.
The results show that the folding codes in the amino acid sequence that dictate the formation of β-strands, α-helices and turns can be deciphered through the evaluation of the hydrophobic interactions among neighbored side-chains of an unfolded polypeptide from a β-strand-like thermodynamic metastable state with great accuracy of prediction. The folding process of a tertiary structure from secondary structures is also involved in the entropy-enthalpy compensation mechanism, since a β-sheet structure can be regarded as a partial tertiary structure. Six other examples are illustrated in Supplementary Materials S2. The folding of secondary structures make hydrophobic side-chains cluster together, thereby inducing thermodynamic pressure on neighbored secondary structures in sequences, which then aggregate or “collapse” into one or more global conformations with one or more hydrophobic cores. This explains why multi-domain proteins sometimes have multiple hydrophobic cores. Enthalpy-entropy compensation may allow some secondary structures folding on the ribosome as this allows certain order of folding of local hydrophobic cores of different domains.
In order to prove that the entropy-enthalpy compensation mechanism is the protein-folding mechanism and can be used to predict the secondary structure of proteins, we preliminarily program a simple software (See Supplementary Materials S5) for predicting the typical secondary structures of α-helices and β-sheets based on the entropy-enthalpy compensation analysis of the amino acid sequences (https://www.researchgate.net/publication/353445795_software, accessed on 30 July 2021) similar to that shown in
Figure 7 and Supplementary Materials S2. Using this software, we successfully identified 5837 of the samples are basically β-strands and α-helices, covering about 96 percent of all those β-strands and 92 percent α-helices in the 1000 proteins (see Supplementary Materials S3). Only 0.5% samples are neither β-strands not α-helices. Hydrophobic effects can most likely contribute to the formation of α-helices through implementing the hydrophobic interaction among neighbored side-chains two or three residues intervals. We used this to identify α-helices from these samples. Then, we identified 2308 samples of β-strands of three or more amino acids long, making the successful rate of the prediction about 81%. We also identified 2416 samples of α-helices, making the successful rate of the prediction about 87% (see Supplementary Materials S3). Moreover, physical folding codes for β-strand and α-helices can be quickly deciphered by using the software, making the overall time for prediction for the 1000 proteins less than 30 s by using only one CPU. We used another 1000 experimentally determined small protein structures to test the software. There were 188 similar sequences in the 1000 samples. All the 1000 small proteins were also randomly selected from the PDB. By using the software, we identified 5915 of the samples are basically β-strands and α-helices, covering about 93 percent of all those β-strands and α-helices in the 1000 proteins. Another 327 samples (about 0.5%) are false predictions. The successful rate of the prediction for β-strand is about 80% and the successful rate of the prediction for α-helix is about 86% (see Supplementary Materials S4). Lateral hydrogen bonding process of segments of β-strands during the folding process of a β-sheet is driven by hydrophobic interactions among β-strands and therefore the entropy-enthalpy compensations (see
Figure 3 and
Figure 4). Thus, a large β-sheet structure can be regarded as a partial tertiary structure. Our model directly predicted the secondary structures in full-length, that is, different from the assembly pathway captured by the molecular dynamics trajectories (see Supplementary Materials S2)
[21]. By analyzing these 2000 proteins, we found that hydrophobic amino acids account for about 55% of the amino acids in the β-strands, and hydrophobic amino acids account for about 47% of the amino acids in the α-helices. About 95% hydrophobic side-chains in the β-strands are involved in hydrophobic interaction with other hydrophobic side-chains in the secondary structures. About 96% hydrophobic side-chains in the α-helices are involved in hydrophobic interaction with other hydrophobic side-chains in the secondary structures.
The assembling process of tertiary structures into a quaternary structure is likely to be essentially the same as that of protein docking. A recent theoretical study found that the binding affinity between the cellular receptor human angiotensin converting enzyme 2 (ACE2) and receptor-binding domain (RBD) in spike (S) protein of novel severe acute respiratory syndrome coronavirus 2(SARS-CoV-2) is determined by the hydrophobic interaction between them
[12]. The hydrophobic interaction and enthalpy-entropy compensation in the binding region between the S protein and ACE2 protein enable the hydrophilic residues in this region to discard the hydrogen-bonded water molecules, and to promote intermolecular hydrogen bonding and electrostatic attraction among these hydrophilic side-chains at the binding site
[12]. Therefore, the folding of protein quaternary structures should be guided by the entropy-enthalpy compensations in between the docking sites according to the Gibbs free energy equation. Namely, entropy increments caused by hydrophobic surface areas collapse in-between protein subunits compensate the increment of enthalpy caused by H-bonds formation between protein subunits. The distribution of hydrophobic and hydrophilic surface areas at smooth docking sites can be easily analyzed from their projective images (see
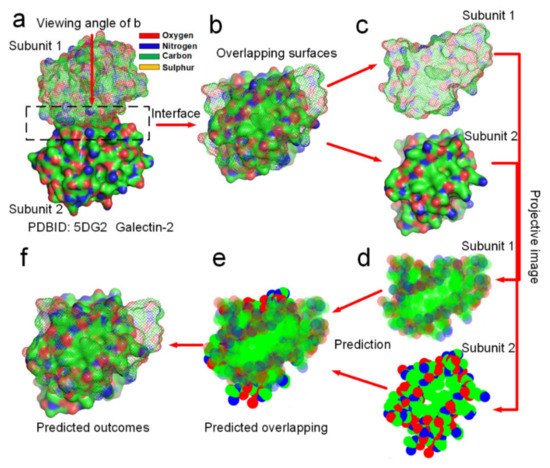
Figure 8). Through analyzing the hydrophobic attraction relationships among proteins of hundreds of dimeric proteins, we find out that the docking position of a dimer is always characterized by two rules of the distribution of hydrophobic and hydrophilic surface areas in their projective images of the overlapping map. First, the docking position maximizes the overlapping of hydrophobic surface areas of the two projective images of the protein subunits. Secondly, subunit–subunit docking sites must allow several hydrogen bond donors and acceptors close to each other in the overlapping position of the two projective images, enabling the formation of several H-bonds between them. Obviously, these two rules conform to the theory that the entropy-enthalpy compensation dominates subunit–subunit docking of dimers into quaternary structures. We had programmed a simple software (https://www.researchgate.net/publication/352552505_software, accessed on 30 July 2021) by using the two rules for predicting the docking position between two projective images of a protein–protein complex. To prove that the folding process from subunit structures into quaternary structures is guided by the entropy-enthalpy compensations, we try to predict the overlapping position of the docking sites of 12 dimers in two dimensions of the projective images (see
Figure 8 and Supplementary Materials S6) by using this software and the two rules of the entropy-enthalpy compensation at the interfaces. By using the software, we find out that the docking position between two projective images of a dimer can be accurate predicted through rotation and translation of the two projective images following the two rules. All the overlapping positions of the docking sites of 12 dimers in two dimensions were successfully predicted by the using the software, which provides potent proof for the entropy-enthalpy compensation theory. All the 12 dimers have relatively smooth binding sites and were randomly selected from the PDB. The docking position between subunit structures indeed maximize the hydrophobic collapse of hydrophobic surface areas of the binding sites in-between the protein subunits.
Figure 8. Prediction of the docking position between two protein subunits of the galectin-2 dimer in two dimensions by using entropy-enthalpy compensation mechanism. (a) The galectin-2 dimer. (b) Distribution of hydrophobic (green areas) and hydrophilic (red and blue areas) surface areas on the two protein subunits at the docking site. (c,d) Projective images of distribution of hydrophobic and hydrophilic surface areas at the binding site. (e) The predicted maximized the overlapping of hydrophobic surface areas of the two projective images of the two protein subunits. (f) The prediction of the docking position between the two protein subunits in two dimensions, almost same as (b).
 +1 credit
+1 credit